13 Techniques of Molecular Genetics
13.1 Polymerase chain reaction
Polymerase chain reaction (PCR) is a method widely used in molecular biology to make copies of a specific DNA segment. Using PCR, copies of DNA sequences are exponentially amplified to generate thousands to millions of more copies of that particular DNA segment. PCR is now a common and often indispensable technique used in medical laboratory and clinical laboratory research for a broad variety of applications including biomedical research and criminal forensics. The vast majority of PCR methods rely on thermal cycling. Thermal cycling exposes reactants to repeated cycles of heating and cooling to permit different temperature-dependent reactions – specifically, DNA melting and enzyme-driven DNA replication.
A basic PCR set-up requires several components and reagents, including a DNA template that contains the DNA target region to amplify; a DNA polymerase; an enzyme that polymerizes new DNA strands; heat-resistant Taq polymerase is especially common, as it is more likely to remain intact during the high-temperature DNA denaturation process; two DNA primers that are complementary to the 3’ (three prime) ends of each of the sense and anti-sense strands of the DNA target (DNA polymerase can only bind to and elongate from a double-stranded region of DNA; without primers there is no double-stranded initiation site at which the polymerase can bind); specific primers that are complementary to the DNA target region are selected beforehand, and are often custom-made in a laboratory or purchased from commercial biochemical suppliers; deoxynucleoside triphosphates, or dNTPs (sometimes called “deoxynucleotide triphosphates”; nucleotides containing triphosphate groups), the building blocks from which the DNA polymerase synthesizes a new DNA strand; a buffer solution providing a suitable chemical environment for optimum activity and stability of the DNA polymerase; bivalent cations, typically magnesium (Mg) or manganese (Mn) ions; Mg2+ is the most common, but Mn2+ can be used for PCR-mediated DNA mutagenesis, as a higher Mn2+ concentration increases the error rate during DNA synthesis; and monovalent cations, typically potassium (K) ions
The reaction is commonly carried out in a volume of 10–200 μL in small reaction tubes (0.2–0.5 mL volumes) in a thermal cycler. The thermal cycler heats and cools the reaction tubes to achieve the temperatures required at each step of the reaction (see below). Many modern thermal cyclers make use of the Peltier effect, which permits both heating and cooling of the block holding the PCR tubes simply by reversing the electric current. Thin-walled reaction tubes permit favorable thermal conductivity to allow for rapid thermal equilibration. Most thermal cyclers have heated lids to prevent condensation at the top of the reaction tube. Older thermal cyclers lacking a heated lid require a layer of oil on top of the reaction mixture or a ball of wax inside the tube.
Almost all PCR applications employ a heat-stable DNA polymerase, such as Taq polymerase, an enzyme originally isolated from the thermophilic bacterium Thermus aquaticus. If the polymerase used was heat-susceptible, it would denature under the high temperatures of the denaturation step. Before the use of Taq polymerase, DNA polymerase had to be manually added every cycle, which was a tedious and costly process.
Applications of the technique include DNA cloning for sequencing, gene cloning and manipulation, gene mutagenesis; construction of DNA-based phylogenies, or functional analysis of genes; diagnosis and monitoring of hereditary diseases; amplification of ancient DNA; analysis of genetic fingerprints for DNA profiling (for example, in forensic science and parentage testing); and detection of pathogens in nucleic acid tests for the diagnosis of infectious diseases.
PCR amplifies a specific region of a DNA strand (the DNA target). Most PCR methods amplify DNA fragments of between 0.1 and 10 kilo base pairs (kbp) in length, although some techniques allow for amplification of fragments up to 40 kbp. The amount of amplified product is determined by the available substrates in the reaction, which become limiting as the reaction progresses.
13.1.1 Procedure
Typically, PCR consists of a series of 20–40 repeated temperature changes, called thermal cycles, with each cycle commonly consisting of two or three discrete temperature steps (see figure below). The cycling is often preceded by a single temperature step at a very high temperature (>90 °C (194 °F)), and followed by one hold at the end for final product extension or brief storage. The temperatures used and the length of time they are applied in each cycle depend on a variety of parameters, including the enzyme used for DNA synthesis, the concentration of bivalent ions and dNTPs in the reaction, and the melting temperature (Tm) of the primers. The individual steps common to most PCR methods are as follows:

- Initialization: This step is only required for DNA polymerases that require heat activation by hot-start PCR. It consists of heating the reaction chamber to a temperature of 94–96 °C (201–205 °F), or 98 °C (208 °F) if extremely thermostable polymerases are used, which is then held for 1–10 minutes.
- Denaturation: This step is the first regular cycling event and consists of heating the reaction chamber to 94–98 °C (201–208 °F) for 20–30 seconds. This causes DNA melting, or denaturation, of the double-stranded DNA template by breaking the hydrogen bonds between complementary bases, yielding two single-stranded DNA molecules.
- Annealing: In the next step, the reaction temperature is lowered to 50–65 °C (122–149 °F) for 20–40 seconds, allowing annealing of the primers to each of the single-stranded DNA templates. Two different primers are typically included in the reaction mixture: one for each of the two single-stranded complements containing the target region. The primers are single-stranded sequences themselves, but are much shorter than the length of the target region, complementing only very short sequences at the 3’ end of each strand. It is critical to determine a proper temperature for the annealing step because efficiency and specificity are strongly affected by the annealing temperature. This temperature must be low enough to allow for hybridization of the primer to the strand, but high enough for the hybridization to be specific, i.e., the primer should bind only to a perfectly complementary part of the strand, and nowhere else. If the temperature is too low, the primer may bind imperfectly. If it is too high, the primer may not bind at all. A typical annealing temperature is about 3–5 °C below the Tm of the primers used. Stable hydrogen bonds between complementary bases are formed only when the primer sequence very closely matches the template sequence. During this step, the polymerase binds to the primer-template hybrid and begins DNA formation.
- Extension/elongation: The temperature at this step depends on the DNA polymerase used; the optimum activity temperature for the thermostable DNA polymerase of Taq (Thermus aquaticus) polymerase is approximately 75–80 °C (167–176 °F), though a temperature of 72 °C (162 °F) is commonly used with this enzyme. In this step, the DNA polymerase synthesizes a new DNA strand complementary to the DNA template strand by adding free dNTPs from the reaction mixture that are complementary to the template in the 5’-to-3’ direction, condensing the 5’-phosphate group of the dNTPs with the 3’-hydroxy group at the end of the nascent (elongating) DNA strand. The precise time required for elongation depends both on the DNA polymerase used and on the length of the DNA target region to amplify. As a rule of thumb, at their optimal temperature, most DNA polymerases polymerize a thousand bases per minute. Under optimal conditions (i.e., if there are no limitations due to limiting substrates or reagents), at each extension/elongation step, the number of DNA target sequences is doubled. With each successive cycle, the original template strands plus all newly generated strands become template strands for the next round of elongation, leading to exponential (geometric) amplification of the specific DNA target region. The processes of denaturation, annealing and elongation constitute a single cycle. Multiple cycles are required to amplify the DNA target to millions of copies. The formula used to calculate the number of DNA copies formed after a given number of cycles is 2n, where n is the number of cycles. Thus, a reaction set for 30 cycles results in 230, or 1073741824, copies of the original double-stranded DNA target region.
- Final elongation: This single step is optional, but is performed at a temperature of 70–74 °C (158–165 °F) (the temperature range required for optimal activity of most polymerases used in PCR) for 5–15 minutes after the last PCR cycle to ensure that any remaining single-stranded DNA is fully elongated.
- Final hold: The final step cools the reaction chamber to 4–15 °C (39–59 °F) for an indefinite time, and may be employed for short-term storage of the PCR products.
A 1971 paper in the Journal of Molecular Biology by Kjell Kleppe [no] and co-workers in the laboratory of Har Gobind Khorana first described a method of using an enzymatic assay to replicate a short DNA template with primers in vitro. However, this early manifestation of the basic PCR principle did not receive much attention at the time and the invention of the polymerase chain reaction in 1983 is generally credited to Kary Mullis.
When Mullis developed the PCR in 1983, he was working in Emeryville, California for Cetus Corporation, one of the first biotechnology companies, where he was responsible for synthesizing short chains of DNA. Mullis has written that he first conceived the idea for PCR while cruising along the Pacific Coast Highway one night in his car. He was playing in his mind with a new way of analyzing changes (mutations) in DNA when he realized that he had instead invented a method of amplifying any DNA region through repeated cycles of duplication driven by DNA polymerase. In Scientific American, Mullis summarized the procedure: “Beginning with a single molecule of the genetic material DNA, the PCR can generate 100 billion similar molecules in an afternoon. The reaction is easy to execute. It requires no more than a test tube, a few simple reagents, and a source of heat.” DNA fingerprinting was first used for paternity testing in 1988.
Mullis was awarded the Nobel Prize in Chemistry in 1993 for his invention, seven years after he and his colleagues at Cetus first put his proposal to practice. Mullis’s 1985 paper with R. K. Saiki and H. A. Erlich, “Enzymatic Amplification of β-globin Genomic Sequences and Restriction Site Analysis for Diagnosis of Sickle Cell Anemia”—the polymerase chain reaction invention (PCR) – was honored by a Citation for Chemical Breakthrough Award from the Division of History of Chemistry of the American Chemical Society in 2017.
Some controversies have remained about the intellectual and practical contributions of other scientists to Mullis’ work, and whether he had been the sole inventor of the PCR principle.
At the core of the PCR method is the use of a suitable DNA polymerase able to withstand the high temperatures of >90 °C (194 °F) required for separation of the two DNA strands in the DNA double helix after each replication cycle. The DNA polymerases initially employed for in vitro experiments presaging PCR were unable to withstand these high temperatures. So the early procedures for DNA replication were very inefficient and time-consuming, and required large amounts of DNA polymerase and continuous handling throughout the process.
The discovery in 1976 of Taq polymerase — a DNA polymerase purified from the thermophilic bacterium, Thermus aquaticus, which naturally lives in hot (50 to 80 °C (122 to 176 °F)) environments such as hot springs—paved the way for dramatic improvements of the PCR method. The DNA polymerase isolated from T. aquaticus is stable at high temperatures remaining active even after DNA denaturation, thus obviating the need to add new DNA polymerase after each cycle. This allowed an automated thermocycler-based process for DNA amplification.
13.2 DNA Sequencing
DNA sequencing is the process of determining the nucleic acid sequence – the order of nucleotides in DNA. It includes any method or technology that is used to determine the order of the four bases: adenine, guanine, cytosine, and thymine. The advent of rapid DNA sequencing methods has greatly accelerated biological and medical research and discovery.
Knowledge of DNA sequences has become indispensable for basic biological research, and in numerous applied fields such as medical diagnosis, biotechnology, forensic biology, virology and biological systematics. Comparing healthy and mutated DNA sequences can diagnose different diseases including various caners, characterize antibody repertoire, and can be used to guide patient treatment. Having a quick way to sequence DNA allows for faster and more individualized medical care to be administered, and for more organisms to be identified and cataloged.
The rapid speed of sequencing attained with modern DNA sequencing technology has been instrumental in the sequencing of complete DNA sequences, or genomes, of numerous types and species of life, including the human genome and other complete DNA sequences of many animal, plant, and microbial species.
The first full DNA genome to be sequenced was that of bacteriophage φX174 in 1977. Medical Research Council scientists deciphered the complete DNA sequence of the Epstein-Barr virus in 1984, finding it contained 172,282 nucleotides. Completion of the sequence marked a significant turning point in DNA sequencing because it was achieved with no prior genetic profile knowledge of the virus.
The first method for determining DNA sequences involved a location-specific primer extension strategy established by Ray Wu at Cornell University in 1970. DNA polymerase catalysis and specific nucleotide labeling, both of which figure prominently in current sequencing schemes, were used to sequence the cohesive ends of lambda phage DNA. Between 1970 and 1973, Wu, R Padmanabhan and colleagues demonstrated that this method can be employed to determine any DNA sequence using synthetic location-specific primers. Frederick Sanger then adopted this primer-extension strategy to develop more rapid DNA sequencing methods at the MRC Centre, Cambridge, UK and published a method for “DNA sequencing with chain-terminating inhibitors” in 1977. Walter Gilbert and Allan Maxam at Harvard also developed sequencing methods, including one for “DNA sequencing by chemical degradation”. In 1973, Gilbert and Maxam reported the sequence of 24 basepairs using a method known as wandering-spot analysis. Advancements in sequencing were aided by the concurrent development of recombinant DNA technology, allowing DNA samples to be isolated from sources other than viruses.
A non-radioactive method for transferring the DNA molecules of sequencing reaction mixtures onto an immobilizing matrix during electrophoresis was developed by Pohl and co-workers in the early 1980s. Followed by the commercialization of the DNA sequencer “Direct-Blotting-Electrophoresis-System GATC 1500” by GATC Biotech, which was intensively used in the framework of the EU genome-sequencing programme, the complete DNA sequence of the yeast Saccharomyces cerevisiae chromosome II. Leroy E. Hood’s laboratory at the California Institute of Technology announced the first semi-automated DNA sequencing machine in 1986. This was followed by Applied Biosystems’ marketing of the first fully automated sequencing machine, the ABI 370, in 1987 and by Dupont’s Genesis 2000 which used a novel fluorescent labeling technique enabling all four dideoxynucleotides to be identified in a single lane. By 1990, the U.S. National Institutes of Health (NIH) had begun large-scale sequencing trials on Mycoplasma capricolum, Escherichia coli, Caenorhabditis elegans, and Saccharomyces cerevisiae at a cost of US$ 0.75 per base. Meanwhile, sequencing of human cDNA sequences called expressed sequence tags began in Craig Venter’s lab, an attempt to capture the coding fraction of the human genome. In 1995, Venter, Hamilton Smith, and colleagues at The Institute for Genomic Research (TIGR) published the first complete genome of a free-living organism, the bacterium Haemophilus influenzae. The circular chromosome contains 1,830,137 bases and its publication in the journal Science marked the first published use of whole-genome shotgun sequencing, eliminating the need for initial mapping efforts.
By 2001, shotgun sequencing methods had been used to produce a draft sequence of the human genome.
13.2.1 Sanger sequencing
Sanger sequencing is the method which prevailed from the 1980s until the mid-2000s. Over that period, great advances were made in the technique, such as fluorescent labelling, capillary electrophoresis, and general automation. These developments allowed much more efficient sequencing, leading to lower costs. The Sanger method, in mass production form, is the technology which produced the first human genome in 2001, ushering in the age of genomics. However, later in the decade, radically different approaches reached the market, bringing the cost per genome down from US$ 100 million in 2001 to US$ 10,000 in 2011.
Sanger sequencing is a method of DNA sequencing based on the selective incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication. Developed by Frederick Sanger and colleagues in 1977, it was the most widely used sequencing method for approximately 40 years. It was first commercialized by Applied Biosystems in 1986. More recently, higher volume Sanger sequencing has been replaced by “Next-Gen” sequencing methods, especially for large-scale, automated genome analyses. However, the Sanger method remains in wide use, for smaller-scale projects, and for validation of Next-Gen results. It still has the advantage over short-read sequencing technologies (like Illumina) that it can produce DNA sequence reads of > 500 nucleotides.
The classical chain-termination method requires a single-stranded DNA template, a DNA primer, a DNA polymerase, normal deoxynucleotidetriphosphates (dNTPs), and modified di-deoxynucleotidetriphosphates (ddNTPs), the latter of which terminate DNA strand elongation. These chain-terminating nucleotides lack a 3’-OH group required for the formation of a phosphodiester bond between two nucleotides, causing DNA polymerase to cease extension of DNA when a modified ddNTP is incorporated. The ddNTPs may be radioactively or fluorescently labelled for detection in automated sequencing machines.
Technical variations of chain-termination sequencing include tagging with nucleotides containing radioactive phosphorus for radiolabelling, or using a primer labeled at the 5’ end with a fluorescent dye. Dye-primer sequencing facilitates reading in an optical system for faster and more economical analysis and automation. The later development by Leroy Hood and coworkers of fluorescently labeled ddNTPs and primers set the stage for automated, high-throughput DNA sequencing.
Dye-terminator sequencing utilizes labelling of the chain terminator ddNTPs, which permits sequencing in a single reaction, rather than four reactions as in the labelled-primer method. In dye-terminator sequencing, each of the four dideoxynucleotide chain terminators is labelled with fluorescent dyes, each of which emit light at different wavelengths.
Owing to its greater expediency and speed, dye-terminator sequencing is now the mainstay in automated sequencing. Its limitations include dye effects due to differences in the incorporation of the dye-labelled chain terminators into the DNA fragment, resulting in unequal peak heights and shapes in the electronic DNA sequence trace chromatogram after capillary electrophoresis (see figure to the left).
This problem has been addressed with the use of modified DNA polymerase enzyme systems and dyes that minimize incorporation variability, as well as methods for eliminating “dye blobs”. The dye-terminator sequencing method, along with automated high-throughput DNA sequence analyzers, was used for the vast majority of sequencing projects until the introduction of Next Generation Sequencing.
Automated DNA-sequencing instruments (DNA sequencers) can sequence up to 384 DNA samples in a single batch. Batch runs may occur up to 24 times a day. DNA sequencers separate strands by size (or length) using capillary electrophoresis, they detect and record dye fluorescence, and output data as fluorescent peak trace chromatograms. Sequencing reactions (thermocycling and labelling), cleanup and re-suspension of samples in a buffer solution are performed separately, before loading samples onto the sequencer. A number of commercial and non-commercial software packages can trim low-quality DNA traces automatically. These programs score the quality of each peak and remove low-quality base peaks (which are generally located at the ends of the sequence). The accuracy of such algorithms is inferior to visual examination by a human operator, but is adequate for automated processing of large sequence data sets.
Chain-termination methods have greatly simplified DNA sequencing. For example, chain-termination-based kits are commercially available that contain the reagents needed for sequencing, pre-aliquoted and ready to use. Limitations include non-specific binding of the primer to the DNA, affecting accurate read-out of the DNA sequence, and DNA secondary structures affecting the fidelity of the sequence.
The DNA sample is divided into four separate sequencing reactions, containing all four of the standard deoxynucleotides (dATP, dGTP, dCTP and dTTP) and the DNA polymerase. To each reaction is added only one of the four dideoxynucleotides (ddATP, ddGTP, ddCTP, or ddTTP), while the other added nucleotides are ordinary ones. The dideoxynucleotide concentration should be approximately 100-fold lower than that of the corresponding deoxynucleotide (e.g. 0.005mM ddTTP : 0.5mM dTTP) to allow enough fragments to be produced while still transcribing the complete sequence. Putting it in a more sensible order, four separate reactions are needed in this process to test all four ddNTPs. Following rounds of template DNA extension from the bound primer, the resulting DNA fragments are heat denatured and separated by size using gel electrophoresis. In the original publication of 1977, the formation of base-paired loops of ssDNA was a cause of serious difficulty in resolving bands at some locations. This is frequently performed using a denaturing polyacrylamide-urea gel with each of the four reactions run in one of four individual lanes (lanes A, T, G, C). The DNA bands may then be visualized by autoradiography or UV light and the DNA sequence can be directly read off the X-ray film or gel image.
13.2.2 High-throughput sequencing (HTS) methods
Several new methods for DNA sequencing were developed in the mid to late 1990s and were implemented in commercial DNA sequencers by the year 2000. Together these were called the “next-generation” or “second-generation” sequencing (NGS) methods, in order to distinguish them from the aforementioned earlier methods, like Sanger Sequencing. In contrast to the first generation of sequencing, NGS technology is typically characterized by being highly scalable, allowing the entire genome to be sequenced at once. Usually, this is accomplished by fragmenting the genome into small pieces, randomly sampling for a fragment, and sequencing it using one of a variety of technologies, such as those described below. An entire genome is possible because multiple fragments are sequenced at once (giving it the name “massively parallel” sequencing) in an automated process.
The high demand for low-cost sequencing has driven the development of high-throughput sequencing technologies that parallelize the sequencing process, producing thousands or millions of sequences concurrently. High-throughput sequencing technologies are intende to lower the cost of DNA sequencing beyond what is possible with standard dye-terminator methods. In ultra-high-throughput sequencing as many as 500,000 sequencing-by-synthesis operations may be run in parallel. Such technologies led to the ability to sequence an entire human genome in as little as one day. As of 2019, corporate leaders in the development of high-throughput sequencing products included Illumina, Qiagen and ThermoFisher Scientific.
13.2.3 454 pyrosequencing
A parallelized version of pyrosequencing was developed by 454 Life Sciences, which has since been acquired by Roche Diagnostics. The method amplifies DNA inside water droplets in an oil solution (emulsion PCR), with each droplet containing a single DNA template attached to a single primer-coated bead that then forms a clonal colony. The sequencing machine contains many picoliter-volume wells each containing a single bead and sequencing enzymes. Pyrosequencing uses luciferase to generate light for detection of the individual nucleotides added to the nascent DNA, and the combined data are used to generate sequence reads. This technology provides intermediate read length and price per base compared to Sanger sequencing on one end and Solexa and SOLiD on the other.
13.2.4 Illumina (Solexa) sequencing
Solexa, now part of Illumina, developed a sequencing method based on reversible dye-terminators technology, and engineered polymerases. The reversible terminated chemistry concept was invented by Bruno Canard and Simon Sarfati at the Pasteur Institute in Paris. It was developed internally at Solexa by those named on the relevant patents. In 2004, Solexa acquired the company Manteia Predictive Medicine in order to gain a massively parallel sequencing technology invented in 1997 by Pascal Mayer and Laurent Farinelli. It is based on “DNA clusters” or “DNA colonies”, which involves the clonal amplification of DNA on a surface. The cluster technology was co-acquired with Lynx Therapeutics of California. Solexa Ltd. later merged with Lynx to form Solexa Inc.
In this method, DNA molecules and primers are first attached on a slide or flow cell and amplified with polymerase so that local clonal DNA colonies, later coined “DNA clusters”, are formed. To determine the sequence, four types of reversible terminator bases (RT-bases) are added and non-incorporated nucleotides are washed away. A camera takes images of the fluorescently labeled nucleotides. Then the dye, along with the terminal 3’ blocker, is chemically removed from the DNA, allowing for the next cycle to begin. Unlike pyrosequencing, the DNA chains are extended one nucleotide at a time and image acquisition can be performed at a delayed moment, allowing for very large arrays of DNA colonies to be captured by sequential images taken from a single camera.
Decoupling the enzymatic reaction and the image capture allows for optimal throughput and theoretically unlimited sequencing capacity. With an optimal configuration, the ultimately reachable instrument throughput is thus dictated solely by the analog-to-digital conversion rate of the camera, multiplied by the number of cameras and divided by the number of pixels per DNA colony required for visualizing them optimally (approximately 10 pixels/colony). In 2012, with cameras operating at more than 10 MHz A/D conversion rates and available optics, fluidics and enzymatics, throughput can be multiples of 1 million nucleotides/second, corresponding roughly to 1 human genome equivalent at 1x coverage per hour per instrument, and 1 human genome re-sequenced (at approx. 30x) per day per instrument (equipped with a single camera).
13.3 RNA-Seq
RNA-Seq uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample at a given moment, analyzing the continuously changing cellular transcriptome.
Specifically, RNA-Seq facilitates the ability to look at alternative gene spliced transcripts, post-transcriptional modifications, gene fusion, mutations/SNPs and changes in gene expression over time, or differences in gene expression in different groups or treatments. In addition to mRNA transcripts, RNA-Seq can look at different populations of RNA to include total RNA, small RNA, such as miRNA, tRNA, and ribosomal profiling. RNA-Seq can also be used to determine exon/intron boundaries and verify or amend previously annotated 5’ and 3’ gene boundaries. Recent advances in RNA-Seq include single cell sequencing and in situ sequencing of fixed tissue.
Prior to RNA-Seq, gene expression studies were done with hybridization-based microarrays. Issues with microarrays include cross-hybridization artifacts, poor quantification of lowly and highly expressed genes, and needing to know the sequence a priori. Because of these technical issues, transcriptomics transitioned to sequencing-based methods. These progressed from Sanger sequencing of Expressed Sequence Tag libraries, to chemical tag-based methods (e.g., serial analysis of gene expression), and finally to the current technology, next-gen sequencing of cDNA (notably RNA-Seq).
The general steps to prepare a complementary DNA (cDNA) library for sequencing are:
- RNA Isolation: RNA is isolated from tissue and mixed with deoxyribonuclease (DNase). DNase reduces the amount of genomic DNA. The amount of RNA degradation is checked with gel and capillary electrophoresis and is used to assign an RNA integrity number to the sample. This RNA quality and the total amount of starting RNA are taken into consideration during the subsequent library preparation, sequencing, and analysis steps.
- RNA selection/depletion: To analyze signals of interest, the isolated RNA can either be kept as is, filtered for RNA with 3’ polyadenylated (poly(A)) tails to include only mRNA, depleted of ribosomal RNA (rRNA), and/or filtered for RNA that binds specific sequences (RNA selection and depletion methods table, below). The RNA with 3’ poly(A) tails are mature, processed, coding sequences. Poly(A) selection is performed by mixing RNA with poly(T) oligomers covalently attached to a substrate, typically magnetic beads. Poly(A) selection ignores noncoding RNA and introduces 3’ bias, which is avoided with the ribosomal depletion strategy. The rRNA is removed because it represents over 90% of the RNA in a cell, which if kept would drown out other data in the transcriptome.
- cDNA synthesis: RNA is reverse transcribed to cDNA because DNA is more stable and to allow for amplification (which uses DNA polymerases) and leverage more mature DNA sequencing technology. Amplification subsequent to reverse transcription results in loss of strandedness, which can be avoided with chemical labeling or single molecule sequencing. Fragmentation and size selection are performed to purify sequences that are the appropriate length for the sequencing machine. The RNA, cDNA, or both are fragmented with enzymes, sonication, or nebulizers. Fragmentation of the RNA reduces 5’ bias of randomly primed-reverse transcription and the influence of primer binding sites, with the downside that the 5’ and 3’ ends are converted to DNA less efficiently. Fragmentation is followed by size selection, where either small sequences are removed or a tight range of sequence lengths are selected. Because small RNAs like miRNAs are lost, these are analyzed independently. The cDNA for each experiment can be indexed with a hexamer or octamer barcode, so that these experiments can be pooled into a single lane for multiplexed sequencing.
When sequencing RNA other than mRNA, the library preparation is modified. The cellular RNA is selected based on the desired size range. For small RNA targets, such as miRNA, the RNA is isolated through size selection. This can be performed with a size exclusion gel, through size selection magnetic beads, or with a commercially developed kit. Once isolated, linkers are added to the 3’ and 5’ end then purified. The final step is cDNA generation through reverse transcription.
13.3.1 Single-cell RNA sequencing (scRNA-Seq)
Standard methods such as microarrays and standard bulk RNA-Seq analysis analyze the expression of RNAs from large populations of cells. In mixed cell populations, these measurements may obscure critical differences between individual cells within these populations.
Single-cell RNA sequencing (scRNA-Seq) provides the expression profiles of individual cells. Although it is not possible to obtain complete information on every RNA expressed by each cell, due to the small amount of material available, patterns of gene expression can be identified through gene clustering analyses. This can uncover the existence of rare cell types within a cell population that may never have been seen before.
Current scRNA-Seq protocols involve the following steps: isolation of single cell and RNA, reverse transcription (RT), amplification, library generation and sequencing. Early methods separated individual cells into separate wells; more recent methods encapsulate individual cells in droplets in a microfluidic device, where the reverse transcription reaction takes place, converting RNAs to cDNAs. Each droplet carries a DNA “barcode” that uniquely labels the cDNAs derived from a single cell. Once reverse transcription is complete, the cDNAs from many cells can be mixed together for sequencing; transcripts from a particular cell are identified by the unique barcode.
Challenges for scRNA-Seq include preserving the initial relative abundance of mRNA in a cell and identifying rare transcripts. The reverse transcription step is critical as the efficiency of the RT reaction determines how much of the cell’s RNA population will be eventually analyzed by the sequencer. The processivity of reverse transcriptases and the priming strategies used may affect full-length cDNA production and the generation of libraries biased toward 3’ or 5’ end of genes.
In the amplification step, either PCR or in vitro transcription (IVT) is currently used to amplify cDNA. One of the advantages of PCR-based methods is the ability to generate full-length cDNA. However, different PCR efficiency on particular sequences (for instance, GC content and snapback structure) may also be exponentially amplified, producing libraries with uneven coverage. On the other hand, while libraries generated by IVT can avoid PCR-induced sequence bias, specific sequences may be transcribed inefficiently, thus causing sequence drop-out or generating incomplete sequences. Several scRNA-Seq protocols have been published: Tang et al., STRT, SMART-seq, CEL-seq, RAGE-seq, , Quartz-seq. and C1-CAGE. These protocols differ in terms of strategies for reverse transcription, cDNA synthesis and amplification, and the possibility to accommodate sequence-specific barcodes (i.e. UMIs) or the ability to process pooled samples.
13.4 DNA microarray
A DNA microarray (also commonly known as DNA chip or biochip) is a collection of microscopic DNA spots attached to a solid surface. Scientists use DNA microarrays to measure the expression levels of large numbers of genes simultaneously or to genotype multiple regions of a genome. Each DNA spot contains picomoles (10−12 moles) of a specific DNA sequence, known as probes (or reporters or oligos). These can be a short section of a gene or other DNA element that are used to hybridize a cDNA or cRNA (also called anti-sense RNA) sample (called target) under high-stringency conditions. Probe-target hybridization is usually detected and quantified by detection of fluorophore-, silver-, or chemiluminescence-labeled targets to determine relative abundance of nucleic acid sequences in the target. The original nucleic acid arrays were macro arrays approximately 9 cm × 12 cm and the first computerized image based analysis was published in 1981. It was invented by Patrick O. Brown.
The core principle behind microarrays is hybridization between two DNA strands, the property of complementary nucleic acid sequences to specifically pair with each other by forming hydrogen bonds between complementary nucleotide base pairs. A high number of complementary base pairs in a nucleotide sequence means tighter non-covalent bonding between the two strands. After washing off non-specific bonding sequences, only strongly paired strands will remain hybridized. Fluorescently labeled target sequences that bind to a probe sequence generate a signal that depends on the hybridization conditions (such as temperature), and washing after hybridization. Total strength of the signal, from a spot (feature), depends upon the amount of target sample binding to the probes present on that spot. Microarrays use relative quantitation in which the intensity of a feature is compared to the intensity of the same feature under a different condition, and the identity of the feature is known by its position.
13.5 Recombinant DNA
Recombinant DNA (rDNA) molecules are DNA molecules formed by laboratory methods of genetic recombination (such as molecular cloning) to bring together genetic material from multiple sources, creating sequences that would not otherwise be found in the genome.
Recombinant DNA is the general name for a piece of DNA that has been created by combining at least two strands. Recombinant DNA is possible because DNA molecules from all organisms share the same chemical structure, and differ only in the nucleotide sequence within that identical overall structure. Recombinant DNA molecules are sometimes called chimeric DNA, because they can be made of material from two different species, like the mythical chimera. R-DNA technology uses palindromic sequences and leads to the production of sticky and blunt ends.
The DNA sequences used in the construction of recombinant DNA molecules can originate from any species. For example, plant DNA may be joined to bacterial DNA, or human DNA may be joined with fungal DNA. In addition, DNA sequences that do not occur anywhere in nature may be created by the chemical synthesis of DNA, and incorporated into recombinant molecules. Using recombinant DNA technology and synthetic DNA, literally any DNA sequence may be created and introduced into any of a very wide range of living organisms.
Proteins that can result from the expression of recombinant DNA within living cells are termed recombinant proteins. When recombinant DNA encoding a protein is introduced into a host organism, the recombinant protein is not necessarily produced. Expression of foreign proteins requires the use of specialized expression vectors and often necessitates significant restructuring by foreign coding sequences.
Recombinant DNA differs from genetic recombination in that the former results from artificial methods in the test tube, while the latter is a normal biological process that results in the remixing of existing DNA sequences in essentially all organisms.
Molecular cloning is the laboratory process used to create recombinant DNA. It is one of two most widely used methods, along with polymerase chain reaction (PCR), used to direct the replication of any specific DNA sequence chosen by the experimentalist. There are two fundamental differences between the methods. One is that molecular cloning involves replication of the DNA within a living cell, while PCR replicates DNA in the test tube, free of living cells. The other difference is that cloning involves cutting and pasting DNA sequences, while PCR amplifies by copying an existing sequence.
Formation of recombinant DNA requires a cloning vector, a DNA molecule that replicates within a living cell. Vectors are generally derived from plasmids or viruses, and represent relatively small segments of DNA that contain necessary genetic signals for replication, as well as additional elements for convenience in inserting foreign DNA, identifying cells that contain recombinant DNA, and, where appropriate, expressing the foreign DNA. The choice of vector for molecular cloning depends on the choice of host organism, the size of the DNA to be cloned, and whether and how the foreign DNA is to be expressed. The DNA segments can be combined by using a variety of methods, such as restriction enzyme/ligase cloning or Gibson assembly.
In standard cloning protocols, the cloning of any DNA fragment essentially involves seven steps: (1) Choice of host organism and cloning vector, (2) Preparation of vector DNA, (3) Preparation of DNA to be cloned, (4) Creation of recombinant DNA, (5) Introduction of recombinant DNA into the host organism, (6) Selection of organisms containing recombinant DNA, and (7) Screening for clones with desired DNA inserts and biological properties.
Following transplantation into the host organism, the foreign DNA contained within the recombinant DNA construct may or may not be expressed. That is, the DNA may simply be replicated without expression, or it may be transcribed and translated and a recombinant protein is produced. Generally speaking, expression of a foreign gene requires restructuring the gene to include sequences that are required for producing an mRNA molecule that can be used by the host’s translational apparatus (e.g. promoter, translational initiation signal, and transcriptional terminator). Specific changes to the host organism may be made to improve expression of the ectopic gene. In addition, changes may be needed to the coding sequences as well, to optimize translation, make the protein soluble, direct the recombinant protein to the proper cellular or extracellular location, and stabilize the protein from degradation.
In most cases, organisms containing recombinant DNA have apparently normal phenotypes. That is, their appearance, behavior and metabolism are usually unchanged, and the only way to demonstrate the presence of recombinant sequences is to examine the DNA itself, typically using a polymerase chain reaction (PCR) test. Significant exceptions exist, and are discussed below.
If the rDNA sequences encode a gene that is expressed, then the presence of RNA and/or protein products of the recombinant gene can be detected, typically using RT-PCR or western hybridization methods. Gross phenotypic changes are not the norm, unless the recombinant gene has been chosen and modified so as to generate biological activity in the host organism. Additional phenotypes that are encountered include toxicity to the host organism induced by the recombinant gene product, especially if it is over-expressed or expressed within inappropriate cells or tissues.
In some cases, recombinant DNA can have deleterious effects even if it is not expressed. One mechanism by which this happens is insertional inactivation, in which the rDNA becomes inserted into a host cell’s gene. In some cases, researchers use this phenomenon to “knock out” genes to determine their biological function and importance. Another mechanism by which rDNA insertion into chromosomal DNA can affect gene expression is by inappropriate activation of previously unexpressed host cell genes. This can happen, for example, when a recombinant DNA fragment containing an active promoter becomes located next to a previously silent host cell gene, or when a host cell gene that functions to restrain gene expression undergoes insertional inactivation by recombinant DNA.
Recombinant DNA is widely used in biotechnology, medicine and research. Today, recombinant proteins and other products that result from the use of DNA technology are found in essentially every western pharmacy, physician or veterinarian office, medical testing laboratory, and biological research laboratory. In addition, organisms that have been manipulated using recombinant DNA technology, as well as products derived from those organisms, have found their way into many farms, supermarkets, home medicine cabinets, and even pet shops, such as those that sell GloFish and other genetically modified animals.
The most common application of recombinant DNA is in basic research, in which the technology is important to most current work in the biological and biomedical sciences. Recombinant DNA is used to identify, map and sequence genes, and to determine their function. rDNA probes are employed in analyzing gene expression within individual cells, and throughout the tissues of whole organisms. Recombinant proteins are widely used as reagents in laboratory experiments and to generate antibody probes for examining protein synthesis within cells and organisms.
Many additional practical applications of recombinant DNA are found in industry, food production, human and veterinary medicine, agriculture, and bioengineering. Some specific examples are:
- Recombinant human insulin: almost completely replaced insulin obtained from animal sources (e.g. pigs and cattle) for the treatment of insulin-dependent diabetes. A variety of different recombinant insulin preparations are in widespread use. Recombinant insulin is synthesized by inserting the human insulin gene into E. coli, or yeast (Saccharomyces cerevisiae) which then produces insulin for human use. Administered to patients whose pituitary glands generate insufficient quantities to support normal growth and development. Before recombinant HGH became available, HGH for therapeutic use was obtained from pituitary glands of cadavers. This unsafe practice led to some patients developing Creutzfeldt–Jakob disease. Recombinant HGH eliminated this problem, and is now used therapeutically. It has also been misused as a performance-enhancing drug by athletes and others. DrugBank entry
- Recombinant blood clotting factor VIII: a blood-clotting protein that is administered to patients with forms of the bleeding disorder hemophilia, who are unable to produce factor VIII in quantities sufficient to support normal blood coagulation. Before the development of recombinant factor VIII, the protein was obtained by processing large quantities of human blood from multiple donors, which carried a very high risk of transmission of blood borne infectious diseases, for example HIV and hepatitis B. DrugBank entry
- Recombinant hepatitis B vaccine: Hepatitis B infection is controlled through the use of a recombinant hepatitis B vaccine, which contains a form of the hepatitis B virus surface antigen that is produced in yeast cells. The development of the recombinant subunit vaccine was an important and necessary development because hepatitis B virus, unlike other common viruses such as polio virus, cannot be grown in vitro. Vaccine information from Hepatitis B Foundation
- Diagnosis of infection with HIV: each of the three widely used methods for diagnosing HIV infection has been developed using recombinant DNA. The antibody test (ELISA or western blot) uses a recombinant HIV protein to test for the presence of antibodies that the body has produced in response to an HIV infection. The DNA test looks for the presence of HIV genetic material using reverse transcription polymerase chain reaction (RT-PCR). Development of the RT-PCR test was made possible by the molecular cloning and sequence analysis of HIV genomes. HIV testing page from US Centers for Disease Control (CDC)
- Golden rice: a recombinant variety of rice that has been engineered to express the enzymes responsible for β-carotene biosynthesis. This variety of rice holds substantial promise for reducing the incidence of vitamin A deficiency in the world’s population. Golden rice is not currently in use, pending the resolution of regulatory and intellectual property issues.
- Herbicide-resistant crops: commercial varieties of important agricultural crops (including soy, maize/corn, sorghum, canola, alfalfa and cotton) have been developed that incorporate a recombinant gene that results in resistance to the herbicide glyphosate (trade name Roundup), and simplifies weed control by glyphosate application. These crops are in common commercial use in several countries.
- Insect-resistant crops: bacillus thuringeiensis is a bacterium that naturally produces a protein (Bt toxin) with insecticidal properties. The bacterium has been applied to crops as an insect-control strategy for many years, and this practice has been widely adopted in agriculture and gardening. Recently, plants have been developed that express a recombinant form of the bacterial protein, which may effectively control some insect predators. Environmental issues associated with the use of these transgenic crops have not been fully resolved.
The first publications describing the successful production and intracellular replication of recombinant DNA appeared in 1972 and 1973, from Stanford and UCSF. In 1980, Paul Berg, a professor in the Biochemistry Department at Stanford and an author on one of the first papers was awarded the Nobel Prize in Chemistry for his work on nucleic acids “with particular regard to recombinant DNA”. Werner Arber, Hamilton Smith, and Daniel Nathans shared the 1978 Nobel Prize in Physiology or Medicine for the discovery of restriction endonucleases which are used rDNA technology.
Stanford University applied for a US patent on recombinant DNA in 1974, listing the inventors as Herbert W. Boyer and Stanley N. Cohen; this patent was awarded in 1980. The first licensed drug generated using recombinant DNA technology was human insulin, developed by Genentech and licensed by Eli Lilly and Company.
Scientists associated with the initial development of recombinant DNA methods recognized that the potential existed for organisms containing recombinant DNA to have undesirable or dangerous properties. At the 1975 Asilomar Conference on Recombinant DNA, these concerns were discussed and a voluntary moratorium on recombinant DNA research was initiated for experiments that were considered particularly risky. This moratorium was widely observed until the National Institutes of Health (USA) developed and issued formal guidelines for rDNA work. Today, recombinant DNA molecules and recombinant proteins are usually not regarded as dangerous. However, concerns remain about some organisms that express recombinant DNA, particularly when they leave the laboratory and are introduced into the environment or food chain.
13.6 Molecular cloning
Molecular cloning is a set of experimental methods in molecular biology that are used to assemble recombinant DNA molecules and to direct their replication within host organisms. The use of the word cloning refers to the fact that the method involves the replication of one molecule to produce a population of cells with identical DNA molecules. Molecular cloning generally uses DNA sequences from two different organisms: the species that is the source of the DNA to be cloned, and the species that will serve as the living host for replication of the recombinant DNA. Molecular cloning methods are central to many contemporary areas of modern biology and medicine.

In a conventional molecular cloning experiment, the DNA to be cloned is obtained from an organism of interest, then treated with enzymes in the test tube to generate smaller DNA fragments. Subsequently, these fragments are then combined with vector DNA to generate recombinant DNA molecules. The recombinant DNA is then introduced into a host organism (typically an easy-to-grow, benign, laboratory strain of E. coli bacteria). This will generate a population of organisms in which recombinant DNA molecules are replicated along with the host DNA. Because they contain foreign DNA fragments, these are transgenic or genetically modified microorganisms (GMO). This process takes advantage of the fact that a single bacterial cell can be induced to take up and replicate a single recombinant DNA molecule. This single cell can then be expanded exponentially to generate a large amount of bacteria, each of which contain copies of the original recombinant molecule. Thus, both the resulting bacterial population, and the recombinant DNA molecule, are commonly referred to as “clones”. Strictly speaking, recombinant DNA refers to DNA molecules, while molecular cloning refers to the experimental methods used to assemble them. The idea arose that different DNA sequences could be inserted into a plasmid and that these foreign sequences would be carried into bacteria and digested as part of the plasmid. That is, these plasmids could serve as cloning vectors to carry genes.
Virtually any DNA sequence can be cloned and amplified, but there are some factors that might limit the success of the process. Examples of the DNA sequences that are difficult to clone are inverted repeats, origins of replication, centromeres and telomeres. Another characteristic that limits chances of success is large size of DNA sequence. Inserts larger than 10kbp have very limited success, but bacteriophages such as bacteriophage λ can be modified to successfully insert a sequence up to 40 kbp.
Prior to the 1970s, the understanding of genetics and molecular biology was severely hampered by an inability to isolate and study individual genes from complex organisms. This changed dramatically with the advent of molecular cloning methods. Microbiologists, seeking to understand the molecular mechanisms through which bacteria restricted the growth of bacteriophage, isolated restriction endonucleases, enzymes that could cleave DNA molecules only when specific DNA sequences were encountered. They showed that restriction enzymes cleaved chromosome-length DNA molecules at specific locations, and that specific sections of the larger molecule could be purified by size fractionation. Using a second enzyme, DNA ligase, fragments generated by restriction enzymes could be joined in new combinations, termed recombinant DNA. By recombining DNA segments of interest with vector DNA, such as bacteriophage or plasmids, which naturally replicate inside bacteria, large quantities of purified recombinant DNA molecules could be produced in bacterial cultures. The first recombinant DNA molecules were generated and studied in 1972.
Molecular cloning takes advantage of the fact that the chemical structure of DNA is fundamentally the same in all living organisms. Therefore, if any segment of DNA from any organism is inserted into a DNA segment containing the molecular sequences required for DNA replication, and the resulting recombinant DNA is introduced into the organism from which the replication sequences were obtained, then the foreign DNA will be replicated along with the host cell’s DNA in the transgenic organism.
The polymerase chain reaction (PCR) is a rapid version of molecular cloning without the need for replication of the DNA in a living microorganism, as PCR replicates DNA in an in vitro solution, free of living cells.
13.6.1 Procedures
In standard molecular cloning experiments, the cloning of any DNA fragment essentially involves seven steps: (1) Choice of host organism and cloning vector, (2) Preparation of vector DNA, (3) Preparation of DNA to be cloned, (4) Creation of recombinant DNA, (5) Introduction of recombinant DNA into host organism, (6) Selection of organisms containing recombinant DNA, (7) Screening for clones with desired DNA inserts and biological properties.
Although a very large number of host organisms and molecular cloning vectors are in use, the great majority of molecular cloning experiments begin with a laboratory strain of the bacterium Escherichia coli (E. coli) and a plasmid cloning vector. E. coli and plasmid vectors are in common use because they are technically sophisticated, versatile, widely available, and offer rapid growth of recombinant organisms with minimal equipment. If the DNA to be cloned is exceptionally large (hundreds of thousands to millions of base pairs), then a bacterial artificial chromosome or yeast artificial chromosome vector is often chosen.
Specialized applications may call for specialized host-vector systems. For example, if the experimentalists wish to harvest a particular protein from the recombinant organism, then an expression vector is chosen that contains appropriate signals for transcription and translation in the desired host organism. Alternatively, if replication of the DNA in different species is desired (for example, transfer of DNA from bacteria to plants), then a multiple host range vector (also termed shuttle vector) may be selected. In practice, however, specialized molecular cloning experiments usually begin with cloning into a bacterial plasmid, followed by subcloning into a specialized vector.
Whatever combination of host and vector are used, the vector almost always contains four DNA segments that are critically important to its function and experimental utility:
- DNA replication origin is necessary for the vector (and its linked recombinant sequences) to replicate inside the host organism
- one or more unique restriction endonuclease recognition sites to serves as sites where foreign DNA may be introduced
- a selectable genetic marker gene that can be used to enable the survival of cells that have taken up vector sequences
- a tag gene that can be used to screen for cells containing the foreign DNA
The cloning vector is treated with a restriction endonuclease to cleave the DNA at the site where foreign DNA will be inserted. The restriction enzyme is chosen to generate a configuration at the cleavage site that is compatible with the ends of the foreign DNA (see DNA end). Typically, this is done by cleaving the vector DNA and foreign DNA with the same restriction enzyme, for example EcoRI. Most modern vectors contain a variety of convenient cleavage sites that are unique within the vector molecule (so that the vector can only be cleaved at a single site) and are located within a gene (frequently beta-galactosidase) whose inativation can be used to distinguish recombinant from non-recombinant organisms at a later step in the process. To improve the ratio of recombinant to non-recombinant organisms, the cleaved vector may be treated with an enzyme (alkaline phosphatase) that dephosphorylates the vector ends. Vector molecules with dephosphorylated ends are unable to replicate, and replication can only be restored if foreign DNA is integrated into the cleavage site.
For cloning of genomic DNA, the DNA to be cloned is extracted from the organism of interest. Virtually any tissue source can be used (even tissues from extinct animals), as long as the DNA is not extensively degraded. The DNA is then purified using simple methods to remove contaminating proteins (extraction with phenol), RNA (ribonuclease) and smaller molecules (precipitation and/or chromatography). Polymerase chain reaction (PCR) methods are often used for amplification of specific DNA or RNA (RT-PCR) sequences prior to molecular cloning.
DNA for cloning experiments may also be obtained from RNA using reverse transcriptase (complementary DNA or cDNA cloning), or in the form of synthetic DNA (artificial gene synthesis). cDNA cloning is usually used to obtain clones representative of the mRNA population of the cells of interest, while synthetic DNA is used to obtain any precise sequence defined by the designer. Such a designed sequence may be required when moving genes across genetic codes (for example, from the mitochrondria to the nucleus) or simply for increasing expression via codon optimization.
The purified DNA is then treated with a restriction enzyme to generate fragments with ends capable of being linked to those of the vector. If necessary, short double-stranded segments of DNA (linkers) containing desired restriction sites may be added to create end structures that are compatible with the vector.
The creation of recombinant DNA is in many ways the simplest step of the molecular cloning process. DNA prepared from the vector and foreign source are simply mixed together at appropriate concentrations and exposed to an enzyme (DNA ligase) that covalently links the ends together. This joining reaction is often termed ligation. The resulting DNA mixture containing randomly joined ends is then ready for introduction into the host organism.
DNA ligase only recognizes and acts on the ends of linear DNA molecules, usually resulting in a complex mixture of DNA molecules with randomly joined ends. The desired products (vector DNA covalently linked to foreign DNA) will be present, but other sequences (e.g. foreign DNA linked to itself, vector DNA linked to itself and higher-order combinations of vector and foreign DNA) are also usually present. This complex mixture is sorted out in subsequent steps of the cloning process, after the DNA mixture is introduced into cells.
The DNA mixture, previously manipulated in vitro, is moved back into a living cell, referred to as the host organism. The methods used to get DNA into cells are varied, and the name applied to this step in the molecular cloning process will often depend upon the experimental method that is chosen (e.g. transformation, transduction, transfection, electroporation).
When microorganisms are able to take up and replicate DNA from their local environment, the process is termed transformation, and cells that are in a physiological state such that they can take up DNA are said to be competent. In mammalian cell culture, the analogous process of introducing DNA into cells is commonly termed transfection. Both transformation and transfection usually require preparation of the cells through a special growth regime and chemical treatment process that will vary with the specific species and cell types that are used.
Electroporation uses high voltage electrical pulses to translocate DNA across the cell membrane (and cell wall, if present). In contrast, transduction involves the packaging of DNA into virus-derived particles, and using these virus-like particles to introduce the encapsulated DNA into the cell through a process resembling viral infection. Although electroporation and transduction are highly specialized methods, they may be the most efficient methods to move DNA into cells.
Whichever method is used, the introduction of recombinant DNA into the chosen host organism is usually a low efficiency process; that is, only a small fraction of the cells will actually take up DNA. Experimental scientists deal with this issue through a step of artificial genetic selection, in which cells that have not taken up DNA are selectively killed, and only those cells that can actively replicate DNA containing the selectable marker gene encoded by the vector are able to survive.
When bacterial cells are used as host organisms, the selectable marker is usually a gene that confers resistance to an antibiotic that would otherwise kill the cells, typically ampicillin. Cells harboring the plasmid will survive when exposed to the antibiotic, while those that have failed to take up plasmid sequences will die. When mammalian cells (e.g. human or mouse cells) are used, a similar strategy is used, except that the marker gene (in this case typically encoded as part of the kanMX cassette) confers resistance to the antibiotic Geneticin.
Modern bacterial cloning vectors (e.g. pUC19 and later derivatives including the pGEM vectors) use the blue-white screening system to distinguish colonies (clones) of transgenic cells from those that contain the parental vector (i.e. vector DNA with no recombinant sequence inserted). In these vectors, foreign DNA is inserted into a sequence that encodes an essential part of beta-galactosidase, an enzyme whose activity results in formation of a blue-colored colony on the culture medium that is used for this work. Insertion of the foreign DNA into the beta-galactosidase coding sequence disables the function of the enzyme, so that colonies containing transformed DNA remain colorless (white). Therefore, experimentalists are easily able to identify and conduct further studies on transgenic bacterial clones, while ignoring those that do not contain recombinant DNA.
The total population of individual clones obtained in a molecular cloning experiment is often termed a DNA library. Libraries may be highly complex (as when cloning complete genomic DNA from an organism) or relatively simple (as when moving a previously cloned DNA fragment into a different plasmid), but it is almost always necessary to examine a number of different clones to be sure that the desired DNA construct is obtained. This may be accomplished through a very wide range of experimental methods, including the use of nucleic acid hybridizations, antibody probes, polymerase chain reaction, restriction fragment analysis and/or DNA sequencing.
13.7 Mutagenesis
In molecular biology, mutagenesis is an important laboratory technique whereby DNA mutations are deliberately engineered to produce mutant genes, proteins, strains of bacteria, or other genetically modified organisms. The various constituents of a gene, as well as its regulatory elements and its gene products, may be mutated so that the functioning of a genetic locus, process, or product can be examined in detail. The mutation may produce mutant proteins with interesting properties or enhanced or novel functions that may be of commercial use. Mutant strains may also be produced that have practical application or allow the molecular basis of a particular cell function to be investigated.
A large number of methods for achieving experimental mutagenesis have been developed. Initially, the kind of mutations artificially induced in the laboratory were entirely random; methods allowing for more specific site-directed mutagenesis were introduced later. Since 2013, development of the CRISPR/Cas9 technology, based on a prokaryotic viral defense system, has allowed for the editing or mutagenesis of a genome in vivo.
13.7.1 Random mutagenesis
Early approaches to mutagenesis relied on methods which produced entirely random mutations. In such methods, cells or organisms are exposed to mutagens such as UV radiation or mutagenic chemicals, and mutants with desired characteristics are then selected. Hermann Muller discovered in 1927 that X-rays can cause genetic mutations in fruit flies, and went on to use the mutants he created for his studies in genetics. For Escherichia coli, mutants may be selected first by exposure to UV radiation, then plated onto an agar medium. The colonies formed are then replica-plated, one in a rich medium, another in a minimal medium, and mutants that have specific nutritional requirements can then be identified by their inability to grow in the minimal medium. Similar procedures may be repeated with other types of cells and with different media for selection.
A number of methods for generating random mutations in specific proteins were later developed to screen for mutants with interesting or improved properties. These methods may involve the use of doped nucleotides in oligonucleotide synthesis, or conducting a PCR reaction in conditions that enhance misincorporation of nucleotides (error-prone PCR), for example by reducing the fidelity of replication or using nucleotide analogues. A variation of this method for integrating non-biased mutations in a gene is sequence saturation mutagenesis. PCR products which contain mutation(s) are then cloned into an expression vector and the mutant proteins produced can then be characterised.
In animal studies, alkylating agents such as N-ethyl-N-nitrosourea (ENU) have been used to generate mutant mice. Ethyl methanesulfonate (EMS) is also often used to generate animal and plant mutants.
In a European Union law (as 2001/18 directive), this kind of mutagenesis may be used to produce GMOs but the products are exempted from regulation: no labeling, no evaluation.
13.7.2 Site-directed mutagenesis
Many researchers seek to introduce selected changes to DNA in a precise, site-specific manner. Analogs of nucleotides and other chemicals were first used to generate localized point mutations. Such chemicals include aminopurine, which induces an AT to GC transition, while nitrosoguanidine, bisulfite, and N4-hydroxycytidine may induce a GC to AT transition. These techniques allow specific mutations to be engineered into a protein; however, they are not flexible with respect to the kinds of mutants generated, nor are they as specific as later methods of site-directed mutagenesis and therefore have some degree of randomness.
Current techniques for site-specific mutation commonly involve using pre-fabricated mutagenic oligonucleotides in a primer extension reaction with DNA polymerase. This methods allows for point mutation or deletion or insertion of small stretches of DNA at specific sites. Advances in methodology have made such mutagenesis now a relatively simple and efficient process.
The site-directed approach may be done systematically in such techniques as alanine scanning mutagenesis, whereby residues are systematically mutated to alanine in order to identify residues important to the structure or function of a protein.
13.7.3 Gene synthesis
As the cost of DNA oligonucleotide synthesis falls, artificial synthesis of a complete gene is now a viable method for introducing mutations into a gene. This method allows for extensive mutation at multiple sites, including the complete redesign of the codon usage of a gene to optimise it for a particular organism.
13.8 Gene knockout
A gene knockout (abbreviation: KO) is a genetic technique in which one of an organism’s genes is made inoperative (“knocked out” of the organism). However, KO can also refer to the gene that is knocked out or the organism that carries the gene knockout. Knockout organisms or simply knockouts are used to study gene function, usually by investigating the effect of gene loss. Researchers draw inferences from the difference between the knockout organism and normal individuals.
The KO technique is essentially the opposite of a gene knock-in. Knocking out two genes simultaneously in an organism is known as a double knockout (DKO). Similarly the terms triple knockout (TKO) and quadruple knockouts (QKO) are used to describe three or four knocked out genes, respectively. However, one needs to distinguish between heterozygous and homozygous KOs. In the former, only one of two gene copies (alleles) is knocked out, in the latter both are knocked out.
Knockouts are accomplished through a variety of techniques. Originally, naturally occurring mutations were identified and then gene loss or inactivation had to be established by DNA sequencing or other methods.
Traditionally, homologous recombination was the main method for causing a gene knockout. This method involves creating a DNA construct containing the desired mutation. For knockout purposes, this typically involves a drug resistance marker in place of the desired knockout gene. The construct will also contain a minimum of 2kb of homology to the target sequence. The construct can be delivered to stem cells either through microinjection or electroporation. This method then relies on the cell’s own repair mechanisms to recombine the DNA construct into the existing DNA. This results in the sequence of the gene being altered, and most cases the gene will b translated into a nonfunctional protein, if it is translated at all. However, this is an inefficient process, as homologous recombination accounts for only 10−2 to 10-3 of DNA integrations. Often, the drug selection marker on the construct is used to select for cells in which the recombination event has occurred.e
These stem cells now lacking the gene could be used in vivo, for instance in mice, by inserting them into early embryos. If the resulting chimeric mouse contained the genetic change in their germline, this could then be passed on offspring.
In diploid organisms, which contain two alleles for most genes, and may as well contain several related genes that collaborate in the same role, additional rounds of transformation and selection are performed until every targeted gene is knocked out. Selective breeding may be required to produce homozygous knockout animals.
There are currently three methods in use that involve precisely targeting a DNA sequence in order to introduce a double-stranded break. Once this occurs, the cell’s repair mechanisms will attempt to repair this double stranded break, often through non-homologous end joining (NHEJ), which involves directly ligating the two cut ends together. This may be done imperfectly, therefore sometimes causing insertions or deletions of base pairs, which cause frameshift mutations. These mutations can render the gene in which they occur nonfunctional, thus creating a knockout of that gene. This process is more efficient than homologous recombination, and therefore can be more easily used to create biallelic knockouts.
Zinc-finger nucleases consist of DNA binding domains that can precisely target a DNA sequence. Each zinc finger can recognize codons of a desired DNA sequence, and therefore can be modularly assembled to bind to a particular sequence. These binding domains are coupled with a restriction endonuclease that can cause a double stranded break (DSB) in the DNA. Repair processes may introduce mutations that destroy functionality of the gene.
Transcription activator-like effector nucleases (TALENs) also contain a DNA binding domain and a nuclease that can cleave DNA. The DNA binding region consists of amino acid repeats that each recognize a single base pair of the desired targeted DNA sequence. If this cleavage is targeted to a gene coding region, and NHEJ-mediated repair introduces insertions and deletions, a frameshift mutation often results, thus disrupting function of the gene.
Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 is a method for genome editing that contains a guide RNA complexed with a Cas9 protein. The guide RNA can be engineered to match a desired DNA sequence through simple complementary base pairing, as opposed to the time consuming assembly of constructs required by zinc-fingers or TALENs. The coupled Cas9 will cause a double stranded break in the DNA. Following the same principle as zinc-fingers and TALENs, the attempts to repair these double stranded breaks often result in frameshift mutations that result in an nonfunctional gene.
13.9 Conditional gene knockout
Conditional gene knockout is a technique used to eliminate a specific gene in a certain tissue, such as the liver. This technique is useful to study the role of individual genes in living organisms. It differs from traditional gene knockout because it targets specific genes at specific times rather than being deleted from beginning of life. Using the conditional gene knockout technique eliminates many of the side effects from traditional gene knockout. In traditional gene knockout, embryonic death from a gene mutation can occur, and this prevents scientists from studying the gene in adults. Some tissues cannot be studied properly in isolation, so the gene must be inactive in a certain tissue while remaining active in others. With this technology, scientists are able to knockout genes at a specific stage in development and study how the knockout of a gene in one tissue affects the same gene in other tissues.
The most commonly used technique is the Cre-lox recombination system. The Cre recombinase enzyme specifically recognizes two lox (loci of recombination) sites within DNA and causes recombination between them. During recombination two strands of DNA exchange information. This recombination will cause a deletion or inversion of the genes between the two lox sites, depending on their orientation. An entire gene can be removed to inactivate it. This whole system is inducible so a chemical can be added to knock genes out at a specific time. Two of the most commonly used chemicals are tetracycline, which activates transcription of the Cre recombinase gene and tamoxifen, which activates transport of the Cre recombinase protein to the nucleus. Only a few cell types express Cre recombinase and no mammalian cells express it so there is no risk of accidental activation of lox sites when using conditional gene knockout in mammals. Figuring out how to express Cre-recombinase in an organism tends to be the most difficult part of this technique. Uses The conditional gene knockout method is often used to model human diseases in other mammals. It has increased scientists’ ability to study diseases, such as cancer, that develop in specific cell types or developmental stages. It is known that mutations in the BRCA1 gene are linked to breast cancer. Scientists used conditional gene knockout to delete the BRCA1 allele in mammary gland tissue in mice and found that it plays an important role in tumour suppression.
Conditional gene knockouts in mice are often used to study human diseases because many genes produce similar phenotypes in both species. The goal of KOMP is to create knockout mutations in the embryonic stem cells for each of the 20,000 protein coding genes in mice. The genes are knocked out because this is the best way to study their function and learn more about their role in human diseases. Some alleles in this project cannot be knocked out using traditional methods and require the specificity of the conditional gene knockout technique. Other combinatorial methods are needed to knockout the last remaining alleles. Conditional gene knockout is a time-consuming procedure and there are additional projects focusing on knocking out the remaining mouse genes. The KOMP projected was started in 2006 and is still ongoing.
13.10 Gene knock-in
Gene knockin is similar to gene knockout, but it replaces a gene with another instead of deleting it.
13.11 RNA interference
RNA interference (RNAi) is a biological process in which RNA molecules inhibit gene expression or translation, by neutralizing targeted mRNA molecules. The process of RNAi was referred to as “co-suppression” and “quelling” when observed prior to the knowledge of an RNA-related mechanism. The discovery of RNAi was preceded first by observations of transcriptional inhibition by antisense RNA expressed in transgenic plants, and more directly by reports of unexpected outcomes in experiments performed by plant scientists in the United States and the Netherlands in the early 1990s. The detailed study of each of these seemingly different processes elucidated that the identity of these phenomena were all actually RNAi. Andrew Fire and Craig C. Mello shared the 2006 Nobel Prize in Physiology or Medicine for their work on RNA interference in the nematode worm Caenorhabditis elegans, which they published in 1998. Since the discovery of RNAi and its regulatory potentials, it has become evident that RNAi has immense potential in suppression of desired genes. RNAi is now known as precise, efficient, stable and better than antisense technology for gene suppression. However, antisense RNA produced intracellularly by an expression vector may be developed and find utility as novel therapeutic agents.
Two types of small ribonucleic acid (RNA) molecules – microRNA (miRNA) and small interfering RNA (siRNA) – are central to RNA interference. RNAs are the direct products of genes, and these small RNAs can direct enzyme complexes to degrade messenger RNA (mRNA) molecules and thus decrease their activity by preventing translation, via post-transcriptional gene silencing. Moreover, transcription can be inhibited via the pre-transcriptional silencing mechanism of RNA interference, through which an enzyme complex catalyzes DNA methylation at genomic positions complementary to complexed siRNA or miRNA. RNA interference has an important role in defending cells against parasitic nucleotide sequences – viruses and transposons. It also influences development.
The RNAi pathway is found in many eukaryotes, including animals, and is initiated by the enzyme Dicer, which cleaves long double-stranded RNA (dsRNA) molecules into short double-stranded fragments of ~21 nucleotide siRNAs. Each siRNA is unwound into two single-stranded RNAs (ssRNAs), the passenger strand and the guide strand. The passenger strand is degraded and the guide strand is incorporated into the RNA-induced silencing complex (RISC). The most well-studied outcome is post-transcriptional gene silencing, which occurs when the guide strand pairs with a complementary sequence in a messenger RNA molecule and induces cleavage by Argonaute 2 (Ago2), the catalytic component of the RISC. In some organisms, this process spreads systemically, despite the initially limited molar concentrations of siRNA.

Figure 13.3: Lentiviral delivery of designed shRNAs and the mechanism of RNA interference in mammalian cells.
RNAi is a valuable research tool, both in cell culture and in living organisms, because synthetic dsRNA introduced into cells can selectively and robustly induce suppression of specific genes of interest. RNAi may be used for large-scale screens that systematically shut down each gene in the cell, which can help to identify the components necessary for a particular cellular process or an event such as cell division. The pathway is also used as a practical tool in biotechnology, medicine and insecticides.
RNAi is an RNA-dependent gene silencing process that is controlled by the RNA-induced silencing complex (RISC) and is initiated by short double-stranded RNA molecules in a cell’s cytoplasm, where they interact with the catalytic RISC component argonaute. When the dsRNA is exogenous (coming from infection by a virus with an RNA genome or laboratory manipulations), the RNA is imported directly into the cytoplasm and cleaved to short fragments by Dicer. The initiating dsRNA can also be endogenous (originating in the cell), as in pre-microRNAs expressed from RNA-coding genes in the genome. The primary transcripts from such genes are first processed to form the characteristic stem-loop structure of pre-miRNA in the nucleus, then exported to the cytoplasm. Thus, the two dsRNA pathways, exogenous and endogenous, converge at the RISC.
Exogenous dsRNA initiates RNAi by activating the ribonuclease protein Dicer, which binds and cleaves double-stranded RNAs (dsRNAs) in plants, or short hairpin RNAs (shRNAs) in humans, to produce double-stranded fragments of 20–25 base pairs with a 2-nucleotide overhang at the 3’ end. Bioinformatics studies on the genomes of multiple organisms suggest this length maximizes target-gene specificity and minimizes non-specific effects. These short double-stranded fragments are called small interfering RNAs (siRNAs). These siRNAs are then separated into single strands and integrated into an active RISC, by RISC-Loading Complex (RLC). RLC includes Dicer-2 and R2D2, and is crucial to unite Ago2 and RISC. TATA-binding protein-associated factor 11 (TAF11) assembles the RLC by facilitating Dcr-2-R2D2 tetramerization, which increases the binding affinity to siRNA by 10-fold. Association with TAF11 would convert the R2-D2-Initiator (RDI) complex into the RLC. R2D2 carries tandem double-stranded RNA-binding domains to recognize the thermodynamically stable terminus of siRNA duplexes, whereas Dicer-2 the other less stable extremity. Loading is asymmetric: the MID domain of Ago2 recognizes the thermodynamically stable end of the siRNA. Therefore, the “passenger” (sense) strand whose 5′ end is discarded by MID is ejected, while the saved “guide” (antisense) strand cooperates with AGO to form the RISC.
After integration into the RISC, siRNAs base-pair to their target mRNA and cleave it, thereby preventing it from being used as a translation template. Differently from siRNA, a miRNA-loaded RISC complex scans cytoplasmic mRNAs for potential complementarity. Instead of destructive cleavage (by Ago2), miRNAs rather target the 3′ untranslated region (UTR) regions of mRNAs where they typically bind with imperfect complementarity, thus blocking the access of ribosomes for translation.
Exogenous dsRNA is detected and bound by an effector protein, known as RDE-4 in C. elegans and R2D2 in Drosophila, that stimulates dicer activity. The mechanism producing this length specificity is unknown and this protein only binds long dsRNAs.
In C. elegans this initiation response is amplified through the synthesis of a population of ‘secondary’ siRNAs during which the dicer-produced initiating or ‘primary’ siRNAs are used as templates. These ‘secondary’ siRNAs are structurally distinct from dicer-produced siRNAs and appear to be produced by an RNA-dependent RNA polymerase (RdRP).
MicroRNA
MicroRNAs (miRNAs) are genomically encoded non-coding RNAs that help regulate gene expression, particularly during development. The phenomenon of RNA interference, broadly defined, includes the endogenously induced gene silencing effects of miRNAs as well as silencing triggered by foreign dsRNA. Mature miRNAs are structurally similar to siRNAs produced from exogenous dsRNA, but before reaching maturity, miRNAs must first undergo extensive post-transcriptional modification. A miRNA is expressed from a much longer RNA-coding gene as a primary transcript known as a pri-miRNA which is processed, in the cell nucleus, to a 70-nucleotide stem-loop structure called a pre-miRNA by the microprocessor complex. This complex consists of an RNase III enzyme called Drosha and a dsRNA-binding protein DGCR8. The dsRNA portion of this pre-miRNA is bound and cleaved by Dicer to produce the mature miRNA molecule that can be integrated into the RISC complex; thus, miRNA and siRNA share the same downstream cellular machinery. First, viral encoded miRNA was described in EBV. Thereafter, an increasing number of microRNAs have been described in viruses. VIRmiRNA is a comprehensive catalogue covering viral microRNA, their targets and anti-viral miRNAs (see also VIRmiRNA resource: http://crdd.osdd.net/servers/virmirna/).
siRNAs derived from long dsRNA precursors differ from miRNAs in that miRNAs, especially those in animals, typically have incomplete base pairing to a target and inhibit the translation of many different mRNAs with similar sequences. In contrast, siRNAs typically base-pair perfectly and induce mRNA cleavage only in a single, specific target. In Drosophila and C. elegans, miRNA and siRNA are processed by distinct argonaute proteins and dicer enzymes.
Three prime untranslated regions (3’UTRs) of messenger RNAs (mRNAs) often contain regulatory sequences that post-transcriptionally cause RNA interference. Such 3’-UTRs often contain both binding sites for microRNAs (miRNAs) as well as for regulatory proteins. By binding to specific sites within the 3’-UTR, miRNAs can decrease gene expression of various mRNAs by either inhibiting translation or directly causing degradation of the transcript. The 3’-UTR also may have silencer regions that bind repressor proteins that inhibit the expression of a mRNA.
The 3’-UTR often contains microRNA response elements (MREs). MREs are sequences to which miRNAs bind. These are prevalent motifs within 3’-UTRs. Among all regulatory motifs within the 3’-UTRs (e.g. including silencer regions), MREs make up about half of the motifs.
As of 2014, the miRBase web site, an archive of miRNA sequences and annotations, listed 28,645 entries in 233 biologic species. Of these, 1,881 miRNAs were in annotated human miRNA loci. miRNAs were predicted to have an average of about four hundred target mRNAs (affecting expression of several hundred genes). Friedman et al. estimate that >45,000 miRNA target sites within human mRNA 3’UTRs are conserved above background levels, and >60% of human protein-coding genes have been under selective pressure to maintain pairing to miRNAs.
Direct experiments show that a single miRNA can reduce the stability of hundreds of unique mRNAs. Other experiments show that a single miRNA may repress the production of hundreds of proteins, but that this repression often is relatively mild (less than 2-fold).
The effects of miRNA dysregulation of gene expression seem to be important in cancer. For instance, in gastrointestinal cancers, nine miRNAs have been identified as epigenetically altered and effective in down regulating DNA repair enzymes.
The effects of miRNA dysregulation of gene expression also seem to be important in neuropsychiatric disorders, such as schizophrenia, bipolar disorder, major depression, Parkinson’s disease, Alzheimer’s disease and autism spectrum disorders.
RISC activation and catalysis Exogenous dsRNA is detected and bound by an effector protein, known as RDE-4 in C. elegans and R2D2 in Drosophila, that stimulates dicer activity. This protein only binds long dsRNAs, but the mechanism producing this length specificity is unknown. This RNA-binding protein then facilitates the transfer of cleaved siRNAs to the RISC complex.
In C. elegans this initiation response is amplified through the synthesis of a population of ‘secondary’ siRNAs during which the dicer-produced initiating or ‘primary’ siRNAs are used as templates. These ‘secondary’ siRNAs are structurally distinct from dicer-produced siRNAs and appear to be produced by an RNA-dependent RNA polymerase (RdRP).
The active components of an RNA-induced silencing complex (RISC) are endonucleases called argonaute proteins, which cleave the target mRNA strand complementary to their bound siRNA. As the fragments produced by dicer are double-stranded, they could each in theory produce a functional siRNA. However, only one of the two strands, which is known as the guide strand, binds the argonaute protein and directs gene silencing. The other anti-guide strand or passenger strand is degraded during RISC activation. Although it was first believed that an ATP-dependent helicase separated these two strands, the process proved to be ATP-independent and performed directly by the protein components of RISC. However, an in vitro kinetic analysis of RNAi in the presence and absence of ATP showed that ATP may be required to unwind and remove the cleaved mRNA strand from the RISC complex after catalysis. The guide strand tends to be the one whose 5’ end is less stably paired to its complement, but strand selection is unaffected by the direction in which dicer cleaves the dsRNA before RISC incorporation. Instead, the R2D2 protein may serve as the differentiating factor by binding the more-stable 5’ end of the passenger strand.
13.11.1 Transcriptional silencing
Components of the RNAi pathway are used in many eukaryotes in the maintenance of the organization and structure of their genomes. Modification of histones and associated induction of heterochromatin formation serves to downregulate genes pre-transcriptionally; this process is referred to as RNA-induced transcriptional silencing (RITS), and is carried out by a complex of proteins called the RITS complex. In fission yeast this complex contains argonaute, a chromodomain protein Chp1, and a protein called Tas3 of unknown function. As a consequence, the induction and spread of heterochromatic regions requires the argonaute and RdRP proteins. Indeed, deletion of these genes in the fission yeast S. pombe disrupts histone methylation and centromere formation, causing slow or stalled anaphase during cell division. In some cases, similar processes associated with histone modification have been observed to transcriptionally upregulate genes.
The type of RNA editing that is most prevalent in higher eukaryotes converts adenosine nucleotides into inosine in dsRNAs via the enzyme adenosine deaminase (ADAR). It was originally proposed in 2000 that the RNAi and A→I RNA editing pathways might compete for a common dsRNA substrate. Some pre-miRNAs do undergo A→I RNA editing and this mechanism may regulate the processing and expression of mature miRNAs. Furthermore, at least one mammalian ADAR can sequester siRNAs from RNAi pathway components. Further support for this model comes from studies on ADAR-null C. elegans strains indicating that A→I RNA editing may counteract RNAi silencing of endogenous genes and transgenes.
13.11.2 Gene knockdown
The RNA interference pathway is often exploited in experimental biology to study the function of genes in cell culture and in vivo in model organisms. Double-stranded RNA is synthesized with a sequence complementary to a gene of interest and introduced into a cell or organism, where it is recognized as exogenous genetic material and activates the RNAi pathway. Using this mechanism, researchers can cause a drastic decrease in the expression of a targeted gene. Studying the effects of this decrease can show the physiological role of the gene product. Since RNAi may not totally abolish expression of the gene, this technique is sometimes referred as a “knockdown”, to distinguish it from “knockout” procedures in which expression of a gene is entirely eliminated. In a recent study validation of RNAi silencing efficiency using gene array data showed 18.5% failure rate across 429 independent experiments.
Extensive efforts in computational biology have been directed toward the design of successful dsRNA reagents that maximize gene knockdown but minimize “off-target” effects. Off-target effects arise when an introduced RNA has a base sequence that can pair with and thus reduce the expression of multiple genes. Such problems occur more frequently when the dsRNA contains repetitive sequences. It has been estimated from studying the genomes of humans, C. elegans and S. pombe that about 10% of possible siRNAs have substantial off-target effects. A multitude of software tools have been developed implementing algorithms for the design of general mammal-specific, and virus-specific siRNAs that are automatically checked for possible cross-reactivity.
Depending on the organism and experimental system, the exogenous RNA may be a long strand designed to be cleaved by dicer, or short RNAs designed to serve as siRNA substrates. In most mammalian cells, shorter RNAs are used because long double-stranded RNA molecules induce the mammalian interferon response, a form of innate immunity that reacts nonspecifically to foreign genetic material. Mouse oocytes and cells from early mouse embryos lack this reaction to exogenous dsRNA and are therefore a common model system for studying mammalian gene-knockdown effects. Specialized laboratory techniques have also been developed to improve the utility of RNAi in mammalian systems by avoiding the direct introduction of siRNA, for example, by stable transfection with a plasmid encoding the appropriate sequence from which siRNAs can be transcribed, or by more elaborate lentiviral vector systems allowing the inducible activation or deactivation of transcription, known as conditional RNAi.
Most functional genomics applications of RNAi in animals have used C. elegans and Drosophila, as these are the common model organisms in which RNAi is most effective. C. elegans is particularly useful for RNAi research for two reasons: firstly, the effects of gene silencing are generally heritable, and secondly because delivery of the dsRNA is extremely simple. Through a mechanism whose details are poorly understood, bacteria such as E. coli that carry the desired dsRNA can be fed to the worms and will transfer their RNA payload to the worm via the intestinal tract. This “delivery by feeding” is just as effective at inducing gene silencing as more costly and time-consuming delivery methods, such as soaking the worms in dsRNA solution and injecting dsRNA into the gonads. Although delivery is more difficult in most other organisms, efforts are also underway to undertake large-scale genomic screening applications in cell culture with mammalian cells.
Functional genomics using RNAi is a particularly attractive technique for genomic mapping and annotation in plants because many plants are polyploid, which presents substantial challenges for more traditional genetic engineering methods. For example, RNAi has been successfully used for functional genomics studies in bread wheat (which is hexaploid) as well as more common plant model systems Arabidopsis and maize.
13.12 Cre-Lox recombination
Cre-Lox recombination is a site-specific recombinase technology, used to carry out deletions, insertions, translocations and inversions at specific sites in the DNA of cells. It allows the DNA modification to be targeted to a specific cell type or be triggered by a specific external stimulus. It is implemented both in eukaryotic and prokaryotic systems. The Cre-lox recombination system has been particularly useful to help neuroscientists to study the brain in which complex cell types and neural circuits come together to generate cognition and behaviors. NIH Blueprint for Neuroscience Research has created several hundreds of Cre driver mouse lines which are currently used by the worldwide neuroscience community.
The system consists of a single enzyme, Cre recombinase, that recombines a pair of short target sequences called the Lox sequences. This system can be implemented without inserting any extra supporting proteins or sequences. The Cre enzyme and the original Lox site called the LoxP sequence are derived from bacteriophage P1.
Placing Lox sequences appropriately allows genes to be activated, repressed, or exchanged for other genes. At a DNA level many types of manipulations can be carried out. The activity of the Cre enzyme can be controlled so that it is expressed in a particular cell type or triggered by an external stimulus like a chemical signal or a heat shock. These targeted DNA changes are useful in cell lineage tracing and when mutants are lethal if expressed globally.
The Cre-Lox system is very similar in action and in usage to the FLP-FRT recombination system.
Cre-Lox recombination involves the targeting of a specific sequence of DNA and splicing it with the help of an enzyme called Cre recombinase. Cre-Lox recombination is commonly used to circumvent embryonic lethality caused by systemic inactivation of many genes. In addition, Cre–Lox recombination provides the best experimental control that presently[when?] exists in transgenic animal modeling to link genotypes (alterations in genomic DNA) to phenotypes (the biological outcomes).
The Cre-lox system is used as a genetic tool to control site specific recombination events in genomic DNA. This system has allowed researchers to manipulate a variety of genetically modified organisms to control gene expression, delete undesired DNA sequences and modify chromosome architecture.
The Cre protein is a site-specific DNA recombinase that can catalyse the recombination of DNA between specific sites in a DNA molecule. These sites, known as loxP sequences, contain specific binding sites for Cre that surround a directional core sequence where recombination can occur.
When cells that have loxP sites in their genome express Cre, a recombination event can occur between the loxP sites. Cre recombinase proteins bind to the first and last 13 bp regions of a lox site forming a dimer. This dimer then binds to a dimer on another lox site to form a tetramer. Lox sites are directional and the two sites joined by the tetramer are parallel in orientation. The double stranded DNA is cut at both loxP sites by the Cre protein. The strands are then rejoined with DNA ligase in a quick and efficient process. The result of recombination depends on the orientation of the loxP sites. For two lox sites on the same chromosome arm, inverted loxP sites will cause an inversion of the intervening DNA, while a direct repeat of loxP sites will cause a deletion event. If loxP sites are on different chromosomes it is possible for translocation events to be catalysed by Cre induced recombination. Two plasmids can be joined using the variant lox sites 71 and 66.
The Cre protein (encoded by the locus originally named as “Causes recombination”, with “Cyclization recombinase” being found in some references) consists of 4 subunits and two domains: The larger carboxyl (C-terminal) domain, and smaller amino (N-terminal) domain. The total protein has 343 amino acids. The C domain is similar in structure to the domain in the Integrase family of enzymes isolated from lambda phage. This is also the catalytic site of the enzyme.
loxP (locus of X-over P1) is a site on the bacteriophage P1 consisting of 34 bp. The site includes an asymmetric 8 bp sequence, variable except for the middle two bases, in between two sets of symmetric, 13 bp sequences. The exact sequence is given below; ‘N’ indicates bases which may vary, and lowercase letters indicate bases that have been mutated from the wild-type. The 13 bp sequences are palindromic but the 8 bp spacer is not, thus giving the loxP sequence a certain direction. Usually loxP sites come in pairs for genetic manipulation. If the two loxP sites are in the same orientation, the floxed sequence (sequence flanked by two loxP sites) is excised; however if the two loxP sites are in the opposite orientation, the floxed sequence is inverted. If there exists a floxed donor sequence, the donor sequence can be swapped with the original sequence. This technique is called recombinase-mediated cassette exchange and is a very convenient and time-saving way for genetic manipulation. The caveat, however, is that the recombination reaction can happen backwards, rendering cassette exchange inefficient. In addition, sequence excision can happen in trans instead of a in cis cassette exchange event. The loxP mutants are created to avoid these problems.
The P1 phage is a temperate phage that causes either a lysogenic or lytic cycle when it infects a bacterium. In its lytic state, once its viral genome is injected into the host cell, viral proteins are produced, virions are assembled, and the host cell is lysed to release the phages, continuing the cycle. In the lysogenic cycle the phage genome replicates with the rest of the bacterial genome and is transmitted to daughter cells at each subsequent cell division. It can transition to the lytic cycle by a later event such as UV radiation or starvation.
Phages like the lambda phage use their site specific recombinases to integrate their DNA into the host genome during lysogeny. P1 phage DNA on the other hand, exists as a plasmid in the host. The Cre-lox system serves several functions in the phage: it circularizes the phage DNA into a plasmid, separates interlinked plasmid rings so they are passed to both daughter bacteria equally and may help maintain copy numbers through an alternative means of replication.
The P1 phage DNA when released into the host from the virion is in the form of a linear double stranded DNA molecule. The Cre enzyme targets loxP sites at the ends of this molecule and cyclises the genome. This can also take place in the absence of the Cre lox system with the help of other bacterial and viral proteins. The P1 plasmid is relatively large (≈90Kbp) and hence exists in a low copy number - usually one per cell. If the two daughter plasmids get interlinked one of the daughter cells of the host will lose the plasmid. The Cre-lox recombination system prevents these situations by unlinking the rings of DNA by carrying out two recombination events (linked rings -> single fused ring -> two unlinked rings). It is also proposed that rolling circle replication followed by recombination will allow the plasmid to increase its copy number when certain regulators (repA) are limiting.
13.13 CRISPR gene editing
CRISPR gene editing is a method by which the genomes of living organisms may be edited. It is based on a simplified version of the bacterial CRISPR/Cas (CRISPR-Cas9) antiviral defense system. By delivering the Cas9 nuclease complexed with a synthetic guide RNA (gRNA) into a cell, the cell’s genome can be cut at a desired location, allowing existing genes to be removed and/or new ones added.
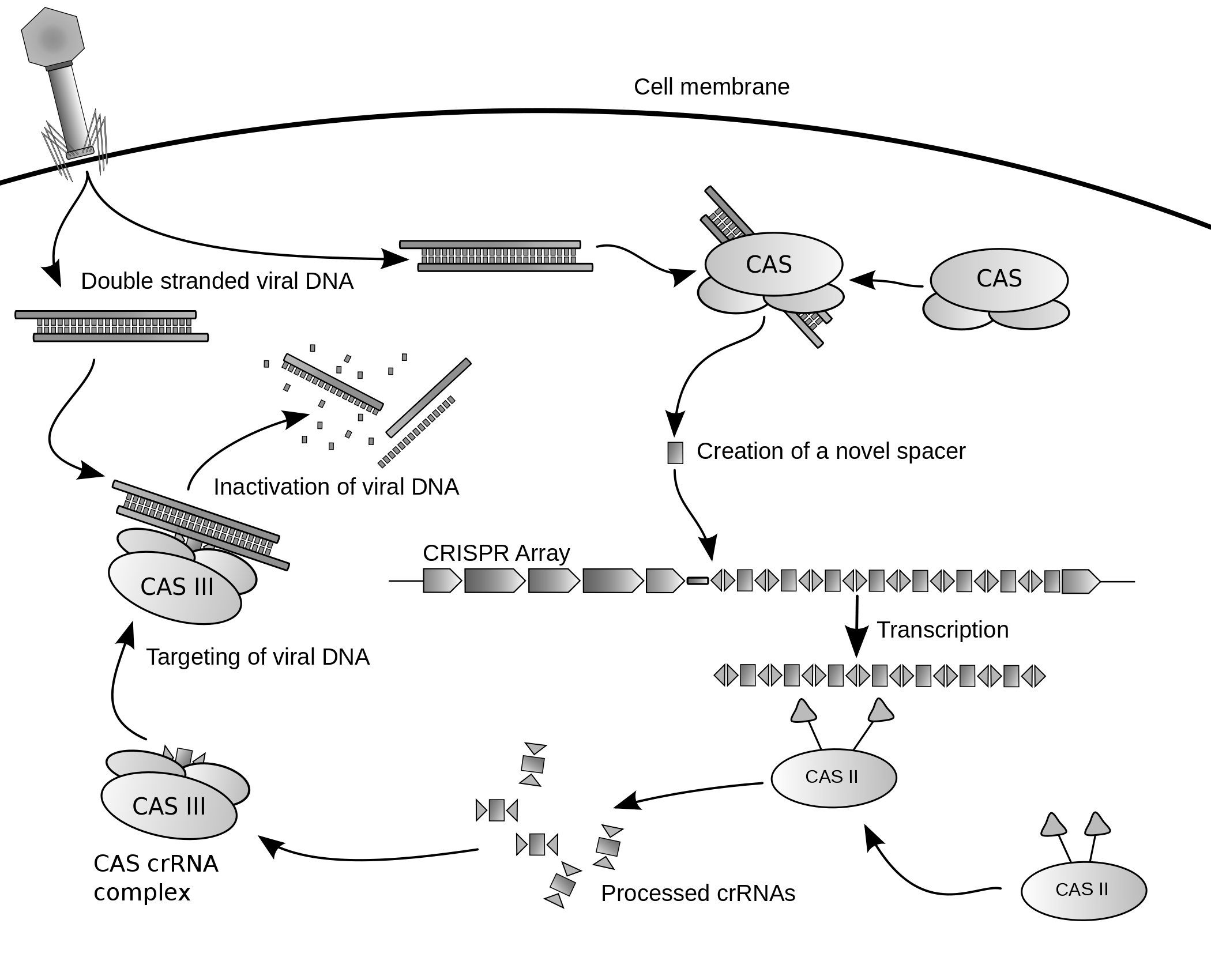
{kind=link}
{kind=link}
{kind=link}
{kind=link}
While genomic editing in eukaryotic cells has been possible using various methods since the 1980s, the methods employed had proved to be inefficient and impractical to implement on a larger scale. Genomic editing leads to irreversible changes to the gene. Working like genetic scissors, the Cas9 nuclease opens both strands of the targeted sequence of DNA to introduce the modification by one of two methods. Knock-in mutations, facilitated via Homology Directed Repair (HDR), is the traditional pathway of targeted genomic editing approaches. This allows for the introduction of targeted DNA damage and repair. HDR employs the use of similar DNA sequences to drive the repair of the break via the incorporation of exogenous DNA to function as the repair template. This method relies on the periodic and isolated occurrence of DNA damage at the target site in order for a repair to commence. Knock-out mutations caused by CRISPR-Cas9 results in the repair of the double-strand break by means of NHEJ (Non-Homologous End Joining). NHEJ can often result in random deletions or insertions at the repair site disrupting or altering gene functionality. Therefore, genomic engineering by CRISPR-Cas9 allows researchers the ability to generate targeted random gene disruption.
Because of this, the precision of genomic editing is a great concern. With the discovery of CRISPR and specifically the Cas9 nuclease molecule, efficient and highly selective editing is now a reality. Cas9 allows for a reliable method of creating a targeted break at a specific location as designated by the crRNA and tracrRna guide strands. Cas9 derived from Streptococcus pyogenes bacteria has facilitated the targeted genomic modification in eukaryotic cells. The ease with which researchers can insert Cas9 and template RNA in order to silence or cause point mutations on specific loci has proved invaluable to the quick and efficient mapping of genomic models and biological processes associated with various genes in a variety of eukaryotes. Newly engineered variants of the Cas9 nuclease have been developed that significantly reduce off-target activity.
CRISPR-Cas9 genome editing techniques have many potential applications, including medicine and crop seed enhancement. The use of CRISPR-Cas9-gRNA complex for genome editing was the AAAS’s choice for breakthrough of the year in 2015. Bioethical concerns have been raised about the prospect of using CRISPR for germline editing.
In the early 2000s, researchers developed zinc finger nucleases (ZFNs), synthetic proteins whose DNA-binding domains enable them to create double-stranded breaks in DNA at specific points. In 2010, synthetic nucleases called transcription activator-like effector nucleases (TALENs) provided an easier way to target a double-stranded break to a specific location on the DNA strand. Both zinc finger nucleases and TALENs require the creation of a custom protein for each targeted DNA sequence, which is a more difficult and time-consuming process than that for guide RNAs. CRISPRs are much easier to design because the process requires making only a short RNA sequence.
Whereas RNA interference (RNAi) does not fully suppress gene function, CRISPR, ZFNs and TALENs provide full irreversible gene knockout. CRISPR can also target several DNA sites simultaneously by simply introducing different gRNAs. In addition, CRISPR costs are relatively low.
CRISPR-Cas9 genome editing is carried out with a Type II CRISPR system. When utilized for genome editing, this system includes Cas9, crRNA, tracrRNA along with an optional section of DNA repair template that is utilized in either non-homologous end joining (NHEJ) or homology directed repair (HDR).
CRISPR-Cas9 often employs a plasmid to transfect the target cells. The main components of this plasmid are displayed in the image and listed in the table. The crRNA needs to be designed for each application as this is the sequence that Cas9 uses to identify and directly bind to the cell’s DNA. The crRNA must bind only where editing is desired. The repair template is designed for each application, as it must overlap with the sequences on either side of the cut and code for the insertion sequence.
Multiple crRNAs and the tracrRNA can be packaged together to form a single-guide RNA (sgRNA). This sgRNA can be joined together with the Cas9 gene and made into a plasmid in order to be transfected into cells.
CRISPR-Cas9 offers a high degree of fidelity and relatively simple construction. It depends on two factors for its specificity: the target sequence and the PAM. The target sequence is 20 bases long as part of each CRISPR locus in the crRNA array. A typical crRNA array has multiple unique target sequences. Cas9 proteins select the correct location on the host’s genome by utilizing the sequence to bond with base pairs on the host DNA. The sequence is not part of the Cas9 protein and as a result is customizable and can be independently synthesized.
The PAM sequence on the host genome is recognized by Cas9. Cas9 cannot be easily modified to recognize a different PAM sequence. However this is not too limiting as it is a short sequence and nonspecific (e.g. the SpCas9 PAM sequence is 5’-NGG-3’ and in the human genome occurs roughly every 8 to 12 base pairs).
Once these have been assembled into a plasmid and transfected into cells the Cas9 protein with the help of the crRNA finds the correct sequence in the host cell’s DNA and – depending on the Cas9 variant – creates a single or double strand break in the DNA.
Properly spaced single strand breaks in the host DNA can trigger homology directed repair, which is less error prone than the non-homologous end joining that typically follows a double strand break. Providing a DNA repair template allows for the insertion of a specific DNA sequence at an exact location within the genome. The repair template should extend 40 to 90 base pairs beyond the Cas9 induced DNA break. The goal is for the cell’s HDR process to utilize the provided repair template and thereby incorporate the new sequence into the genome. Once incorporated, this new sequence is now part of the cell’s genetic material and passes into its daughter cells.
Many online tools are available to aid in designing effective sgRNA sequences.
Delivery of Cas9, sgRNA, and associated complexes into cells can occur via viral and non-viral systems. Electroporation of DNA, RNA, or ribonucleocomplexes is a common technique, though it can result in harmful effects on the target cells. Chemical transfection techniques utilizing lipids have also been used to introduce sgRNA in complex with Cas9 into cells. Hard-to-transfect cells (e.g. stem cells, neurons, and hematopoietic cells) require more efficient delivery systems such as those based on lentivirus (LVs), adenovirus (AdV) and adeno-associated virus (AAV).
Several variants of CRISPR-Cas9 allow gene activation or genome editing with an external trigger such as light or small molecules. These include photoactivatable CRISPR systems developed by fusing light-responsive protein partners with an activator domain and a dCas9 for gene activation, or fusing similar light responsive domains with two constructs of split-Cas9, or by incorporating caged unnatural amino acids into Cas9, or by modifying the guide RNAs with photocleavable complements for genome editing.
Methods to control genome editing with small molecules include an allosteric Cas9, with no detectable background editing, that will activate binding and cleavage upon the addition of 4-hydroxytamoxifen (4-HT), 4-HT responsive intein-linked Cas9s or a Cas9 that is 4-HT responsive when fused to four ERT2 domains. Intein-inducible split-Cas9 allows dimerization of Cas9 fragments and Rapamycin-inducible split-Cas9 system developed by fusing two constructs of split Cas9 with FRB and FKBP fragments. Furthermore, other studies have shown to induce transcription of Cas9 with a small molecule, doxycycline. Small molecules can also be used to improve Homology Directed Repair (HDR), often by inhibiting the Non-Homologous End Joining (NHEJ) pathway. These systems allow conditional control of CRISPR activity for improved precision, efficiency and spatiotemporal control.
Cas9 genomic modification has allowed for the quick and efficient generation of transgenic models within the field of genetics. Cas9 can be easily introduced into the target cells via plasmid transfection along with sgRNA in order to model the spread of diseases and the cell’s response and defense to infection. The ability of Cas9 to be introduced in vivo allows for the creation of more accurate models of gene function, mutation effects, all while avoiding the off-target mutations typically observed with older methods of genetic engineering. The CRISPR and Cas9 revolution in genomic modeling doesn’t only extend to mammals. Traditional genomic models such as Drosophila melanogaster, one of the first model species, have seen further refinement in their resolution with the use of Cas9. Cas9 uses celtimicrobial therapy and a strategy by which to manipulate bacterial populations. Recent studies suggested a correlation between the interfering of the CRISPR-Cas locus and acquisition of antibiotic resistance This system provides protection of bacteria against invading foreign DNA, such as transposons, bacteriophages and plasmids. This system was shown to be a strong selective pressure for the acquisition of antibiotic resistance and virulence factor in bacterial pathogens.
The first clinical trial involving CRISPR started in 2016. It involved removing immune cells from people with lung cancer, using CRISPR to edit out the gene expressed PD-1, then administrating the altered cells back to the same person. 20 other trials were under way or nearly ready, mostly in China, as of 2017.
In 2016, the United States Food and Drug Administration (FDA) approved a clinical trial in which CRISPR would be used to alter T cells extracted from people with different kinds of cancer and then administer those engineered T cells back to the same people.
Knockdown/activation
Using “dead” versions of Cas9 (dCas9) eliminates CRISPR’s DNA-cutting ability, while preserving its ability to target desirable sequences. Multiple groups added various regulatory factors to dCas9s, enabling them to turn almost any gene on or off or adjust its level of activity. Like RNAi, CRISPR interference (CRISPRi) turns off genes in a reversible fashion by targeting, but not cutting a site. The targeted site is methylated, epigenetically modifying the gene. This modification inhibits transcription. These precisely placed modifications may then be used to regulate the effects on gene expressions and DNA dynamics after the inhibition of certain genome sequences within DNA. Within the past few years, epigenetic marks in different human cells have been closely researched and certain patterns within the marks have been found to correlate with everything ranging from tumor growth to brain activity. Conversely, CRISPR-mediated activation (CRISPRa) promotes gene transcription. Cas9 is an effective way of targeting and silencing specific genes at the DNA level. In bacteria, the presence of Cas9 alone is enough to block transcription. For mammalian applications, a section of protein is added. Its guide RNA targets regulatory DNA sequences called promoters that immediately precede the target gene.
Cas9 was used to carry synthetic transcription factors that activated specific human genes. The technique achieved a strong effect by targeting multiple CRISPR constructs to slightly different locations on the gene’s promoter.
13.14 Gene drive
A gene drive is a genetic engineering technology that propagates a particular suite of genes throughout a population by altering the probability that a specific allele will be transmitted to offspring from the natural 50% probability. Gene drives can arise through a variety of mechanisms. They have been proposed to provide an effective means of genetically modifying specific populations and entire species.,The technique can employ adding, deleting, disrupting, or modifying genes. Proposed applications include exterminating insects that carry pathogens (notably mosquitoes that transmit malaria, dengue, and zika pathogens), controlling invasive species or eliminating herbicide or pesticide resistance.
In sexually-reproducing species, most genes are present in two copies (which can be the same or different alleles), either one of which has a 50% chance of passing to a descendant. By biasing the inheritance of particular altered genes, synthetic gene drives could spread alterations through a population.
At the molecular level, an endonuclease gene drive works by cutting a chromosome at a specific site that does not encode the drive, inducing the cell to repair the damage by copying the drive sequence onto the damaged chromosome. The cell then has two copies of the drive sequence. The method derives from genome editing techniques and relies on the fact that double strand breaks are most frequently repaired by homologous recombination, (in the presence of a template), rather than non-homologous end joining. To achieve this behavior, endonuclease gene drives consist of two nested elements:
- either a homing endonuclease or a RNA-guided endonuclease (e.g., Cas9 or Cpf1) and its guide RNA, that cuts the target sequence in recipient cells
- a template sequence used by the DNA repair machinery after the target sequence is cut. To achieve the self-propagating nature of gene drives, this repair template contains at least the endonuclease sequence. Because the template must be used to repair a double-strand break at the cutting site, its sides are homologous to the sequences that are adjacent to the cutting site in the host genome. By targeting the gene drive to a gene coding sequence, this gene will be inactivated; additional sequences can be introduced in the gene drive to encode new functions.
As a result, the gene drive insertion in the genome will re-occur in each organism that inherits one copy of the modification and one copy of the wild-type gene. If the gene drive is already present in the egg cell (e.g. when received from one parent), all the gametes of the individual will carry the gene drive (instead of 50% in the case of a normal gene).
Since it can never more than double in frequency with each generation, a gene drive introduced in a single individual typically requires dozens of generations to affect a substantial fraction of a population. Alternatively, releasing drive-containing organisms in sufficient numbers can affect the rest within a few generations; for instance, by introducing it in every thousandth individual, it takes only 12–15 generations to be present in all individuals. Whether a gene drive will ultimately become fixed in a population and at which speed depends on its effect on individuals fitness, on the rate of allele conversion, and on the population structure. In a well mixed population and with realistic allele conversion frequencies (≈90%), population genetics predicts that gene drives get fixed for selection coefficient smaller than 0.3; in other words, gene drives can be used to spread modifications as long as reproductive success is not reduced by more than 30%. This is in contrast with normal genes, which can only spread across large populations if they increase fitness.
13.15 Human germline modification
As of March 2015, multiple groups had announced ongoing research with the intention of laying the foundations for applying CRISPR to human embryos for human germline engineering, including labs in the US, China, and the UK, as well as US biotechnology company OvaScience. Scientists, including a CRISPR co-discoverer, urged a worldwide moratorium on applying CRISPR to the human germline, especially for clinical use. They said “scientists should avoid even attempting, in lax jurisdictions, germline genome modification for clinical application in humans” until the full implications “are discussed among scientific and governmental organizations”. These scientists support further low-level research on CRISPR and do not see CRISPR as developed enough for any clinical use in making heritable changes to humans.
In April 2015, Chinese scientists reported results of an attempt to alter the DNA of non-viable human embryos using CRISPR to correct a mutation that causes beta thalassemia, a lethal heritable disorder. The study had previously been rejected by both Nature and Science in part because of ethical concerns. The experiments resulted in successfully changing only some of the intended genes, and had off-target effects on other genes. The researchers stated that CRISPR is not ready for clinical application in reproductive medicine. In April 2016, Chinese scientists were reported to have made a second unsuccessful attempt to alter the DNA of non-viable human embryos using CRISPR - this time to alter the CCR5 gene to make the embryo HIV resistant.
In December 2015, an International Summit on Human Gene Editing took place in Washington under the guidance of David Baltimore. Members of national scientific academies of America, Britain and China discussed the ethics of germline modification. They agreed to support basic and clinical research under certain legal and ethical guidelines. A specific distinction was made between somatic cells, where the effects of edits are limited to a single individual, versus germline cells, where genome changes could be inherited by descendants. Heritable modifications could have unintended and far-reaching consequences for human evolution, genetically (e.g. gene/environment interactions) and culturally (e.g. Social Darwinism). Altering of gametocytes and embryos to generate inheritable changes in humans was defined to be irresponsible. The group agreed to initiate an international forum to address such concerns and harmonize regulations across countries.
In November 2018, Jiankui He announced that he had edited two human embryos, to attempt to disable the gene for CCR5, which codes for a receptor that HIV uses to enter cells. He said that twin girls, Lulu and Nana, had been born a few weeks earlier. He said that the girls still carried functional copies of CCR5 along with disabled CCR5 (mosaicism) and were still vulnerable to HIV. The work was widely condemned as unethical, dangerous, and premature. An international group of scientists called for a global moratorium on genetically editing human embryos.
Policy regulations for the CRISPR-Cas9 system vary around the globe. In February 2016, British scientists were given permission by regulators to genetically modify human embryos by using CRISPR-Cas9 and related techniques. However, researchers were forbidden from implanting the embryos and the embryos were to be destroyed after seven days.
The US has an elaborate, interdepartmental regulatory system to evaluate new genetically modified foods and crops. For example, the Agriculture Risk Protection Act of 2000 gives the USDA the authority to oversee the detection, control, eradication, suppression, prevention, or retardation of the spread of plant pests or noxious weeds to protect the agriculture, environment and economy of the US. The act regulates any genetically modified organism that utilizes the genome of a predefined “plant pest” or any plant not previously categorized. In 2015, Yinong Yang successfully deactivated 16 specific genes in the white button mushroom, to make them non-browning. Since he had not added any foreign-species (transgenic) DNA to his organism, the mushroom could not be regulated by the USDA under Section 340.2. Yang’s white button mushroom was the first organism genetically modified with the CRISPR-Cas9 protein system to pass US regulation. In 2016, the USDA sponsored a committee to consider future regulatory policy for upcoming genetic modification techniques. With the help of the US National Academies of Sciences, Engineering and Medicine, special interests groups met on April 15 to contemplate the possible advancements in genetic engineering within the next five years and any new regulations that might be needed as a result. The FDA in 2017 proposed a rule that would classify genetic engineering modifications to animals as “animal drugs”, subjecting them to strict regulation if offered for sale, and reducing the ability for individuals and small businesses to make them profitably.