7 DNA Sequence Analysis
In this exercise, we will learn how to work with DNA sequence data. We will learn how to use various online bioinformatics software tools. We will use a DNA sequence that was obtained in a previous experiment. In the experiment, genomic DNA was isolated from an organism and used as template in a polymerase change reaction (PCR). The primers for the PCR were chosen in the coding region of a gene that is commonly used for “barcoding” of biological species belonging to one of the four kingdoms of eykaryotic organisms. The PCR products were sent to a commercial DNA sequencing facility for sequencing by the Sanger method.
7.1 DNA Sequence Files
The sequencing facility sent the results back in the form of two types of files for each sequenced sample: one type of file has the file extension .ab1. Files of this type are binary files and contain DNA sequence information recorded by an Applied Biosystems DNA sequencer and associated software; also known as a electropherogram file or DNA trace file; can be be viewed graphically by a ABI file viewer to analyze and compare DNA sequences. For conveniencem a second file with the file extension .seq or .txt contains the DNA sequence in ASCII (American Standard Code for Information Interchange) format. Files of this type can be read by any program that can read simple text.
The electropherogram of the sequence that we will be using in this exercise is shown in Figure 7.1 below. An electropherogram, or electrophoregram, is a record or chart produced by an automated DNA sequencer used to separate the labled DNA produced in the Sanger sequencing procedure.
- Inspect the electropherograme (electrophoregram) shown in Fig. 7.1.
- Notice peaks of four different colors (green, blue, black and red) corresponding to the bases adenine (A), cytosine (C), guanine (G), and thymine (T) in the sequenced DNA. Narrow, well separated peaks indicate a good signal and high confidence in “calling the base”, broad, overlapping peaks signify low quality data. If the sofware algorithm used cannot decide which base to call, the letter “N” is used.
- Notice how the peaks are broad and overlapping both at the very beginning and end of the sequence. This is typical.
Figure 7.1: The DNA sequence chromatogram obtained from sequencing the PCR products from the unknown organism. This chromatogram was produced using the free and open-source software programs R and Bioconductor package sangerseqR.
7.2 BLAST Search
We will use the BLAST (Basic Local Alignment Tool) program to compare our DNA with all DNA sequences stored in GenBank. GenBank is the NIH genetic sequence database, an annotated collection of all publicly available DNA sequences. GenBank is part of the international nucleotide sequence database collaboration, which comprises the DNA Databank of Japan (DDBJ), the European Nucleotide Archive (ENA), and GenBank at NCBI. these three organizations exchange data on a daily basis.
There are several ways to search and retrieve data from GenBank:
- Search GenBank for sequence identifiers and annotations with entrez nucleotide, which is divided into three divisions:
- corenucleotide (the main collection)
- dbest (expressed sequence tags)
- dbgss (genome survey sequences).
- Search and align GenBank sequences to a query sequence using BLAST (basic local alignment search tool).
We will use BLAST to search te corenucleotide main collection.
- Open a web browser on your computer. If you are reading this in your browser, right click on the highlighted link to open the web site of the U.S. National Library of Medicine in a new tab (fig. 7.2). Otherwise enter the link in your browser manually.

Figure 7.2: The web page of the U.S. National Library of Medicine.
- On the U.S. National Library of Medicine web page (fig. 7.2), click on the right most square with the DNA icon and BLAST (basic local alignment tool) written on it.
- On the newly opened page (fig. 7.3), you will see a row of squares displaying various titles. Click on the left most square that has nucleotide BLAST written on it. A new page will open.

Figure 7.3: The Basic Local Alignment Search Tool (BLAST) start page.
- Here is the text only version of DNA electropherogram shown in Figure 7.1 above. Copy the sequence to your clipboard:
NNNNNNNNNNNNNNNNNNNNNNNNNNNTAANNNANNCTTCTTTTTTACATTTAACAAGATTTTTTNTACATGAATATAAAGAAA
GGTCTAAAAATGAAAAATTGATTTATTCTTTTAATAAAAGAAATAAATTTGTCACATTTTCTTGGAATTTTTTTTATATTTTTG
AATTGGAATTTTTTTTAACTTCTCTTTTGACTAGGTTTNTAAATAATTTAGTTCTATCCAACTTATTCGATCAAATTAATCTAT
TAGAAAAAATAAATAATAATAATAAAAGTCAATATTTTTTATCAGAAAAAAGGATCTATAACCAAAATTCCTGTATTCATTATG
TACGGTATCAAAATCGTTGTATTATGGCTTCGGAAGGCTTTTATTTTCATGATACGAACTGGATATATTTTATATTGAATATTT
GGCAATTTTTTATGCATTTATGGATTCAACCTTTCAGGTTCTCAACAAAGCATTTTCAAAAGCAGAGTTTTTTTTTTCTGGGTT
ATCAATTTGGCAGAGAATCCAAACTGCTAAAAGTTCGATCTATTTCACTTGATAAATCACCTACAATTTATTCACGTTTAAAAA
AAAATCTTTTAAAAACACAAATTGTATATCCAATAGATTTTCTAGCAAAAGAGGGTTTTTGTGATATTTCAGGTTATCCTATTA
GTAGATCGACTTGGACTACNTCGACNGATGAAGAANTTTTATTGAATTTTAATAAAATTTGNAAAAGCTTTTATTTTTATTATG
GGGGGTTGATAAAAAAAGACATTTTATANNNAATCANNTATATCCTCCGATTTTCATGTGCAAAAACNCTANCTCGTAANCNNN
NAANNNNNAANNNNNNA- Paste the copied DNA sequence into the white box on the top left (Fig. 7.4).

Figure 7.4: Nucleotide BLAST (BLASTN) sequence entry form.
- Leave the default values unchanged and click on the oval blue button at the lower left that has BLAST written on it (Fig. 7.5). This will upload your sequence (referred to from now on as the Query sequence) to the server where it will be compared to all sequences on record.

Figure 7.5: After you have pasted your sequence into the sequence entry field, click the BLAST button at the lower left of the page.
- A new page will open that will be updated every 2 seconds while your query is being processed (Fig. 7.6).

Figure 7.6: The BLASTN query status updated page.
- Once the search has completed, the BLASTN results page will open (Fig. 7.7).

Figure 7.7: The BLASTN results page.
- Click on the “+” sign in front of “Graphic Summary” (Fig. 7.7). A graphical summary of your results will be shown (Fig. 7.8. The light green line on top represents your query sequence, below are shown any retreived sequences that align with your sequence.

Figure 7.8: Graphic summary of the BLASTN search results.
- Click on the “+” sign in front of “Description” (Fig. 7.9). A list of sequences that align with your query sequence will be shown (Fig. 7.9.

Figure 7.9: List of sequences producing significant alignments with your query sequence.
- Click on the first sequence. A new page will showing the alignment of the query sequence with the retrieved sequence (Fig. 7.10).

Figure 7.10: Alignment of the best retrieved sequence with the query sequence.
Which sequence matches our query sequence?
Is this sequence linked to a scientific publication? If so, download the original paper.
What is the scientific name of the organism that has a matching sequence? What is the common English name of this organism? What type of organism is it?
What is the name of the gene that matches this sequence? Does the query sequence correspond to a partial or the full-length sequence of this gene?
Go back to the list of sequence alignments produced by BLAST. Look for the sequnce with GenBank accession AY348551.1 (accession identifiers are listed in the left most column). Click on it. A new page will open. 15. On which chromosome is this gene located? How many nucleotides does this chromosome have?
How many amino acids does the full-lenght gene code for?
Find open reading frame (ORF) in the sequence. Write down the nucleotide mumber corresponding to the the first base of the start codon (ATG). Find the stop codon. Write down the number of the last nucleotide of the stop codon. You will need those numbers in the next exercise (“Design of PCR Primers”).
What is the function of this gene in the organism?
Click on the “GenBank” link on the top left of the page and choose “FASTA”.
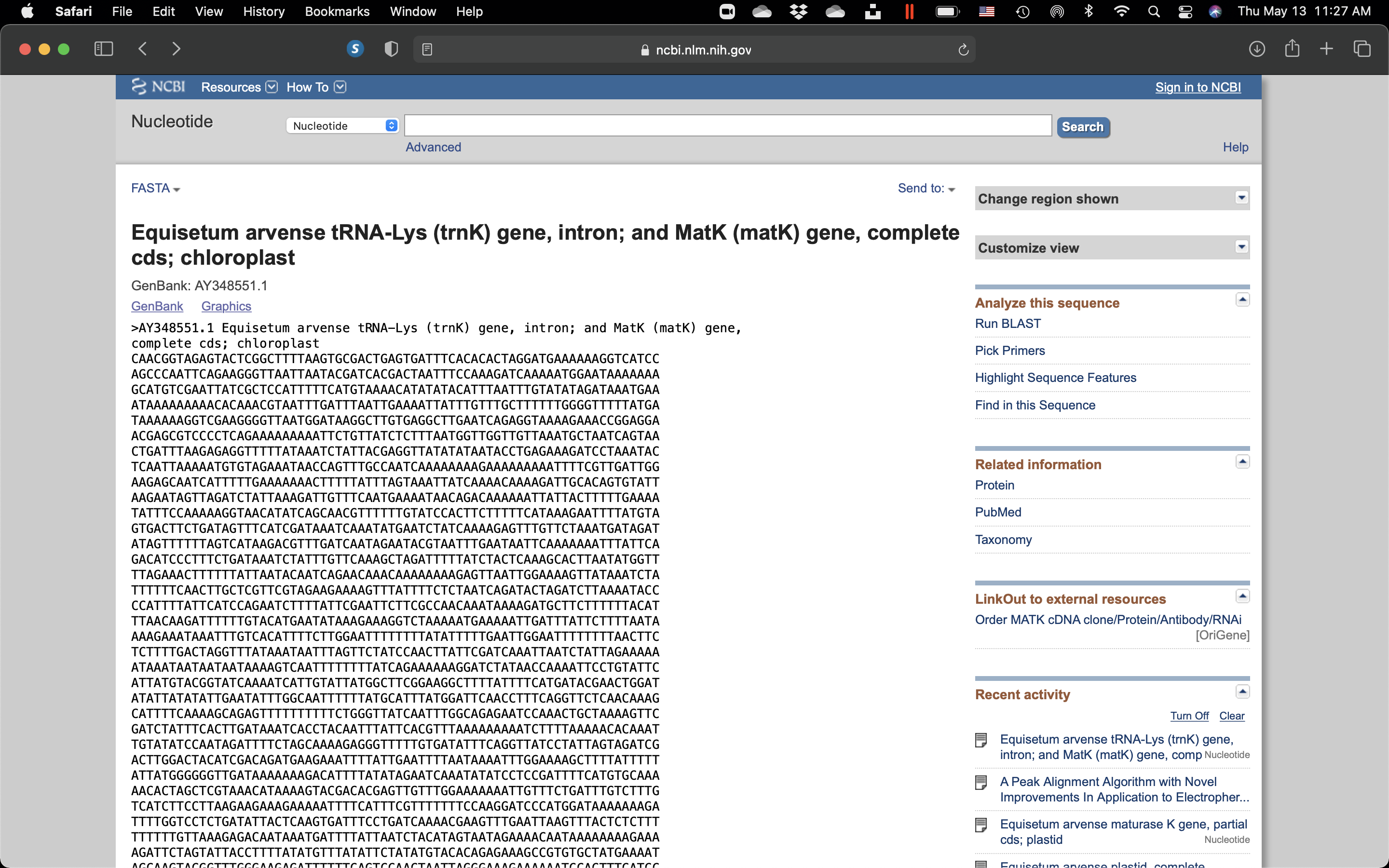
Figure 7.11: View of the DNA seuence with GenBank accession # AY348551.1 in FASTA format.
In bioinformatics and biochemistry, the FASTA format is a text-based format for representing either nucleotide sequences or amino acid (protein) sequences, in which nucleotides or amino acids are represented using single-letter codes. The format also allows for sequence names and comments to precede the sequences. The format originates from the FASTA software package, but has now become a near universal standard in the field of bioinformatics.
In the original format, a sequence was represented as a series of lines, each of which was no longer than 120 characters and usually did not exceed 80 characters. This probably was to allow for preallocation of fixed line sizes in software: at the time most users relied on Digital Equipment Corporation (DEC) VT220 (or compatible) terminals which could display 80 or 132 characters per line.[citation needed] Most people preferred the bigger font in 80-character modes and so it became the recommended fashion to use 80 characters or less (often 70) in FASTA lines. Also, the width of a standard printed page is 70 to 80 characters (depending on the font). Hence, 80 characters became the norm.
The first line in a FASTA file started either with a “>” (greater-than) symbol or, less frequently, a “;” (semicolon) was taken as a comment. Subsequent lines starting with a semicolon would be ignored by software. Since the only comment used was the first, it quickly became used to hold a summary description of the sequence, often starting with a unique library accession number, and with time it has become commonplace to always use “>” for the first line and to not use “;” comments (which would otherwise be ignored).
Following the initial line (used for a unique description of the sequence) was the actual sequence itself in standard one-letter character string. Anything other than a valid character would be ignored (including spaces, tabulators, asterisks, etc…). It was also common to end the sequence with an "*" (asterisk) character (in analogy with use in PIR formatted sequences) and, for the same reason, to leave a blank line between the description and the sequence.
- Towards the top left of the page, just above the title, click on “Send to” (Fig. 7.12), in the pop-up, make sure that “Complete Record” is highlighted, and under “Choose Destination”, click on “File”, then under “Format”, choose “Fasta”, and then click on “Create File”. Save the file to your computer.

Figure 7.12: Save the gene sequence in FASTA format
- Click on the “GenBank” link on the top left of the page and choose “Graphics”. Click on the different buttons and explore the many options and tools available. For example, show the open reading frames in all six frames. Find the open reading frame for the matK protein.

Figure 7.13: Graphics view of the DNA seuence with GenBank accession # AY348551.1.